






心肌梗死后心功能障碍竟然有“乳酸化”的影子
背景
心肌梗死(MI)是由心脏或冠状动脉持续缺血引起的常见表现,并与高死亡率相关。心肌纤维化是病理性细胞外基质重塑和成纤维细胞活化的过程,是包括心肌梗死在内的大多数心脏损伤不可避免的事件。乳酸已被证明可诱导肌成纤维细胞分化并促进肺纤维化。研究发现糖酵解衍生的乳酸通过在组蛋白赖氨酸(K)残基上添加乳酸基直接改变组蛋白,这一过程被称为“乳酸化”,最终影响基因转录。然而,乳酸是否会诱导心肌缺血损伤后的乳酸化和心肌纤维化尚不清楚。
内皮向间充质转化(EndoMT)是内皮细胞经历的一个过程,在这个过程中,一系列细胞和分子的变化导致间充质细胞(如肌成纤维细胞)表型的改变。心肌梗死后的心脏缺氧会促进EndoMT,从而导致心脏纤维化,从而导致心功能障碍。
在这里,作者观察到乳酸水平的增加通过诱导心肌EndoMT而强烈地增强心肌纤维化和心功能障碍。在体外,作者发现乳酸促进缺氧诱导的内皮细胞EndoMT和TGF-β/ Smad2信号的激活。在机制上,作者证明了乳酸在缺氧/心肌梗死后上调Snail1核易位和乳酸化。抑制Snail1可改善心肌梗死后乳酸诱导的心功能障碍,抑制乳酸刺激的EndoMT、Snail1的乳酸化和TGF-β/Smad2信号的激活。这些发现增强了作者对乳酸盐在心肌梗死后EndoMT中的作用的理解,并将成为开发改善心肌梗死后心脏重塑和功能的创新疗法的基础。本文于2023年2月发表于《SCIENCE ADVANCES》,IF=13.6。

技术路线

结果
1.减少乳酸可改善心肌梗死后的心功能和心肌纤维化
临床研究表明,高乳酸水平在心力衰竭患者中普遍存在,并与心力衰竭的死亡率相关。在本研究中,作者研究了心肌梗死是否会增加循环乳酸水平。如图1 (A至F)所示,术后7天,心肌梗死显著提高血清和心脏乳酸水平,同时心功能下降。为了确定乳酸的减少是否会改善心肌梗死后心功能障碍和心肌纤维化过程,作者通过腹腔注射糖酵解抑制剂2-脱氧-d -葡萄糖(2-DG)来降低乳酸水平。2 dg抑制血清和心肌乳酸水平引起的心肌梗死(图1 A和B),感兴趣的射血分数(EF %)和部分缩短(FS %) 2 dg MI老鼠的水平高于汽车MI老鼠(图1C和D)。左心室舒张末期容积(LVEDV)、左室收缩末期容积(LVESV)值比汽车低2 dg MI小鼠心肌梗死小鼠(图1,E和F),表明抑制乳酸生产改善心肌梗死后心脏功能。此外,马森三色染色显示,2-DG给药导致心肌梗死后心肌纤维化减少(图1G)。这些数据表明乳酸生成增加可能参与了MI诱导的纤维化和心功能障碍。

图1. 心肌梗死后乳酸水平升高导致心功能障碍加重和心肌纤维化增加
2.心肌梗死后乳酸生成增加,加重心功能障碍和心肌纤维化
为了证实乳酸在心肌梗死后心功能障碍和纤维化中的作用,作者在心肌梗死诱导后3小时给小鼠补充乳酸,并在心肌梗死后1周和4周用超声心动图评估心功能。图1H显示,腹腔注射乳酸(0.5 g/kg体重)7天后血清乳酸浓度恢复到正常水平。因此,乳酸盐每7天腹腔注射一次。与假手术组相比,心肌梗死显著下调了EF%和FS%的值(图1、I和J)。作者观察到,与心肌梗死组相比,乳酸治疗进一步降低了EF%(7天下降14.0%,28天下降19.5%)和FS%(7天下降15.8%,28天下降21.6%)的值(图1I和J)。与此同时,乳酸治疗增加了心肌梗死后LVEDV和LVESV的水平(图1I和J)。作者还使用渗透性微型泵来持续维持小鼠血清乳酸水平的稳定。乳酸水平的升高显著下调了心肌梗死后EF%和FS%的水平(图B和C),上调了LVEDV和LVESV的值(图D和E)。马松三色染色证明,乳酸给药可显著诱导心肌梗死小鼠的心脏纤维化(图1M)。
3.抑制乳酸生成可减弱心肌梗死后心肌EndoMT
EndoMT是内皮细胞失去其内皮特性并向成纤维细胞样表型过渡的过程,在心肌梗死后的心脏纤维化中起关键作用。为了研究乳酸如何增加心肌梗死后心肌纤维化,作者接下来评估了乳酸与心肌梗死后心肌EndoMT之间是否存在关联。心肌梗死诱导内皮标记物CD31和VE-cadherin表达(图2A)以及间充质标记物成纤维细胞特异性蛋白1 (FSP1)、α-平滑肌肌动蛋白(α-SMA)和胶原1a1表达(图2A)显著增加。然而,通过给药2-DG抑制乳酸生成可提高CD31和VE-cadherin的表达,同时消除心肌梗死诱导的胶原1a1、α-SMA和FSP1的增加(图2A)。与此一致,2-DG处理上调内皮标志物Cdh5和Kdr的mRNA水平(图2B)。相反,2-DG下调了间充质标志物Atca2、Col1a1、Fn1和S100a4 mRNA的表达(图2B)。为了证实作者的观察,作者在内皮细胞特异性绿色荧光蛋白(GFP)标记的小鼠(TIE2GFP)中添加或不添加2-DG诱导心肌梗死,并在心脏组织中使用抗GFP(绿色)和抗fsp1(红色)抗体进行免疫荧光染色。如图2C所示,与假对照组相比,MI诱导GFP与FSP1共定位。相反,2-DG对乳酸生成的抑制减弱了MI诱导的GFP和FSP1之间的共定位,表明2-DG减少了MI刺激的心肌EndoMT。此外,在心肌梗死或假手术后7天,作者从收获的心脏中分离内皮细胞,并通过流式细胞术检测内皮细胞分化为肌成纤维细胞的百分比。如图2D所示,从假对照小鼠分离的内皮细胞显示,大多数GFP细胞被抗cd31和抗cd114(内皮细胞标记物)抗体染色。Gp38是一个被广泛接受的心脏成纤维细胞群体标记。图2E显示,假对照心脏内皮细胞中几乎没有检测到Gp38染色。然而,心肌梗死显著提高了gfp标记的内皮细胞中gp38阳性染色细胞的数量和百分比。相反,2-DG处理抑制了MI诱导的gp38阳性内皮细胞如图2D所示,从假对照小鼠分离的内皮细胞显示,大多数GFP细胞被抗cd31和抗cd114(内皮细胞标记物)抗体染色。Gp38是一个被广泛接受的心脏成纤维细胞群体标记。图2E显示,假对照心脏内皮细胞中几乎没有检测到Gp38染色。然而,心肌梗死显著提高了gfp标记的内皮细胞中gp38阳性染色细胞的数量和百分比。相反,2-DG处理抑制了MI诱导的gp38阳性内皮细胞。
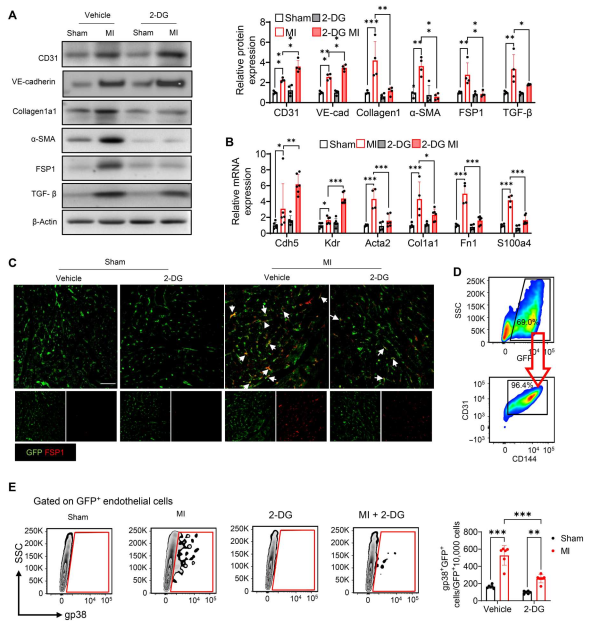
图2. 2-DG可减弱mi诱导的EndoMT
4.高水平的乳酸促进心肌梗死后心肌的EndoMT
然后,作者研究了补充乳酸是否能促进心肌梗死后EndoMT的转变。Western blot显示,与心肌梗死组相比,补充乳酸降低了心肌梗死小鼠内皮细胞标志物(CD31和VE-cadherin)的水平,并促进了间充质标志物(FSP1、α-SMA和Collagen1a1)的表达(图3A)。定量实时聚合酶链反应(qRT-PCR)一致显示,乳酸水平升高导致心肌梗死后Cdh5和Kdr mRNA水平降低(图3B和C),并诱导Col1a1、Fn1和S100a4 mRNA水平(图3、D至F),表明乳酸诱导心肌梗死后EndoMT。接下来,TIE2GFP小鼠接受心肌梗死或假手术,然后给药乳酸。抗GFP(绿色)和抗FSP1(红色)抗体的免疫荧光染色显示,与假小鼠相比,心肌梗死诱导了GFP和FSP1的共定位(图3G)。具有重要意义的是,乳酸处理导致心肌梗死后GFP和FSP1的共定位明显增加(图3G),表明乳酸处理后内皮细胞分化为成纤维细胞的数量增加。同样,流式细胞术显示,乳酸处理进一步上调心肌梗死后GFP+gp38+细胞的数量(图3H),表明乳酸促进心肌梗死后的EndoMT。为了进一步阐明乳酸在心肌梗死后EndoMT中的作用,作者从心肌梗死或假手术小鼠心脏组织中分离内皮细胞。如图5a所示,心肌梗死可促进心脏内皮细胞中胶原1a1、α-SMA和FSP1的表达。乳酸处理进一步刺激心肌梗死后这些间充质标志物的表达。相反,2-DG强烈阻断心肌梗死诱导的胶原1a1、α-SMA和FSP1的表达。
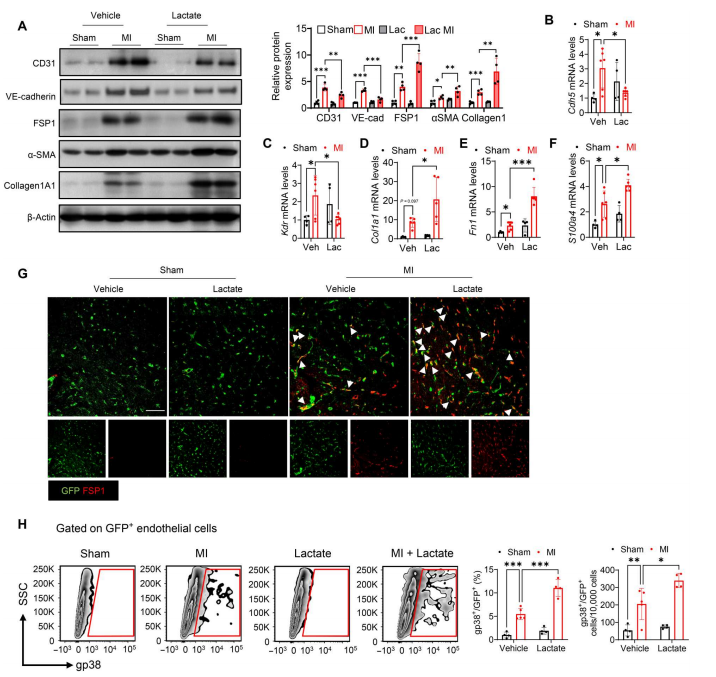
图3. 补充乳酸可促进心肌梗死后的EndoMT
5.乳酸刺激内皮细胞迁移,降低VE-cadherin表达,促进缺氧后的EndoMT
为了确定乳酸促进EndoMT过程的机制,作者使用缺氧刺激的内皮细胞进行了体外实验。众所周知,内皮细胞的迁移和增殖与EndoMT过程有关。因此,作者进行了伤口愈合试验和5-乙基-2-脱氧尿苷(EdU)掺入试验,以观察乳酸是否会改变内皮细胞的迁移和增殖。图4A显示,与正常缺氧对照组相比,缺氧加速了内皮细胞的迁移。此外,在10 mM而不是5 mM处施用乳酸显著增强了缺氧诱导的伤口愈合(图4A),表明乳酸诱导内皮细胞在缺氧刺激后迁移。同样,EdU掺入实验显示乳酸可以促进缺氧刺激的内皮细胞增殖(图4B)。接下来,作者检测了迁移细胞中ve -钙粘蛋白的水平,以揭示乳酸是否会在缺氧后刺激内皮细胞迁移后改变其表型。VE-cadherin免疫荧光染色显示,缺氧72小时后VE-cadherin的完整性受到抑制(图4D)。缺氧后,用10 mM乳酸处理进一步破坏内皮细胞表面ve -钙粘蛋白的完整性(图4D)。此外,Western blot和qRT-PCR分析也显示乳酸处理显著降低了VE-cadherin的水平与低氧组相比(图4C和图5C),表明乳酸可能改变低氧条件下内皮细胞的表型。为了说明乳酸是否可以诱导缺氧后内皮细胞的EndoMT,作者用乳酸处理内皮细胞,并检测缺氧后EndoMT的标志物。如图5A所示,缺氧72小时后内皮细胞形态由典型形态变为细长的纺锤形。与低氧对照相比,乳酸处理导致了更大的细胞形态变化。此外,细长的细胞FSP1染色呈阳性,CD31染色呈阴性(图5B)。图5C显示,低氧刺激后,乳酸降低VE-cadherin表达,诱导α-SMA表达。为了研究乳酸促进的EndoMT是否会诱导内皮细胞功能障碍,作者进行了一项基于matrigel的内皮细胞血管生成实验,发现乳酸可以抑制缺氧诱导的管形成(图5D)。此外,通过胶原凝胶收缩实验,作者证实乳酸处理的内皮细胞在缺氧刺激后表现出间充质样功能,胶原凝胶大小减小,收缩性增加(图5E)。这些数据表明,乳酸诱导缺氧后的EndoMT。
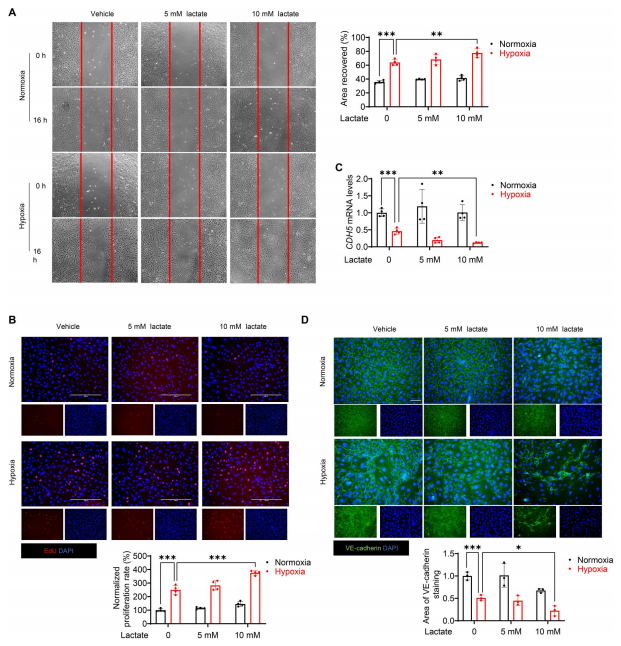
图4. 乳酸诱导内皮细胞迁移,降低缺氧后VE-cadherin的表达

图5. 乳酸促进缺氧后内皮细胞的EndoMT
6.乳酸激活缺氧后TGF-β/Smad2信号
为了确定乳酸诱导缺氧后EndoMT的机制,作者评估了TGF-β和Smad2/3的表达,这两个已知参与调节EndoMT。在体内,qRT-PCR显示,乳酸显著加速心肌梗死后Tgfb1和Smad2 mRNA的表达(图6A和B),但未改变Smad3 mRNA的表达水平(图6C)。此外,
2-DG抑制乳酸生成减轻了MI诱导的Tgfb1和Smad2 mRNA的表达(图6D和E)。体外研究还显示,乳酸促进了缺氧刺激下的人脐带内皮细胞(HUVECs)和HCMECs中Tgfb1和Smad2 mRNA的表达(图6F和G)。与基因表达水平一致,乳酸处理上调Smad2的磷酸化和TGF-β的表达,但不影响缺氧刺激后Smad3的磷酸化(图6H)。综上所述,这些数据表明乳酸诱导的TGF-β/Smad2激活可能有助于缺氧后的EndoMT。

图6. 乳酸刺激心肌梗死/缺氧后TGF-β/Smad2信号传导
7.细胞内乳酸抑制减弱缺氧诱导的EndoMT
单羧酸转运蛋白(mct)是跨质膜转运乳酸的双向转运蛋白。通过在给药前使用MCT抑制剂α-氰基-4-羟基肉桂酸(CHC),作者观察到CHC降低了乳酸处理诱导的细胞内乳酸水平(图7A),表明细胞外乳酸通过MCT转运到内皮细胞中。此外,给药CHC改善了缺氧后乳酸促进的内皮细胞迁移(图7B)。免疫荧光染色显示,CHC阻止乳酸处理降低VE-cadherin表达(图7C),而成管实验显示,CHC促进缺氧后血管生成(图7D)。此外,CHC减轻了乳酸诱导的内皮标志物PECAM1、CDH5和KDR mRNA水平的下降(图7,E至G)以及间质标志物ACTA2、FN1和COL1A1 mRNA水平的升高(图7,H至J)。伴随着这些变化,CHC治疗也抑制了乳酸诱导的TGFB1和SMAD2 mRNA的表达(图7,K和L)。总的来说,这些数据表明细胞外乳酸促进的EndoMT是由mct依赖机制介导的。
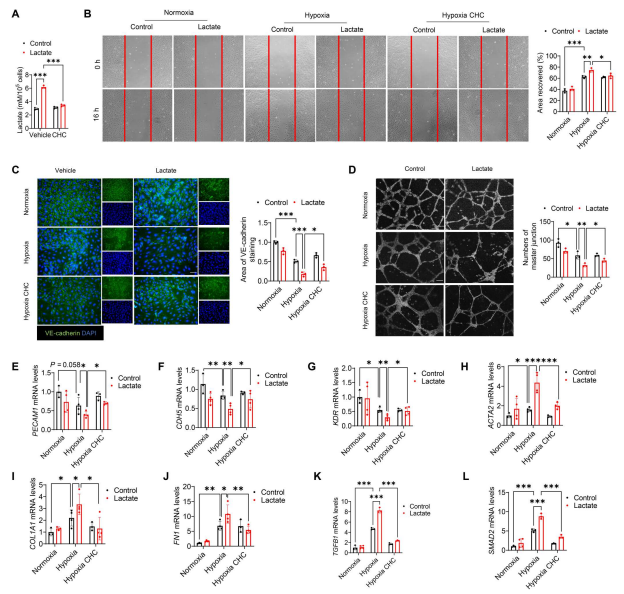
图7. 抑制细胞内乳酸可减弱缺氧诱导的EndoMT
8.乳酸促进缺氧后Snail1核易位
Snail1是TGF-β的转录因子,在调节上皮-间质转化(epithelial-to-mesenchymal transition, EndoMT)中起重要作用。接下来,作者通过TGF-β/ smad2依赖性信号通路研究了Snail1在乳酸诱导的EndoMT中的作用。如图8A所示,单独缺氧不会改变Snail1的细胞质表达。而低氧条件下Snail1核表达明显加快。乳酸处理进一步上调Snail1的核易位。因此,免疫荧光染色显示,低氧刺激后Snail1核染色更加阳性,乳酸可进一步诱导低氧刺激(图8B),表明Snail1核易位可能在乳酸促进的EndoMT中发挥重要作用。然后,作者探讨了Snail1核易位的增加是否与乳酸激活的TGF-β信号传导有关。作者使用抗snail1抗体进行染色质免疫沉淀(ChIP)实验,并检测TGFB1基因是否可以在抗snail1抗体的免疫沉淀中检测到。图8C显示,在缺氧挑战的细胞中,具有TGFB1基因的Snail1大量募集。然而TGFB1基因与Snail1蛋白之间的相互作用在乳酸处理的缺氧挑战细胞中明显大于缺氧细胞(图8C)。这些数据表明,乳酸促进Snail1蛋白和TGFB1基因相互作用,从而促进TGF-β/ smad2介导的EndoMT。

图8. 乳酸促进缺氧后Snail1核易位和乳酸化
9.缺氧后乳酸诱导Snail1乳酸化
表观遗传学在TGF-β相关的EndoMT中起重要作用。最近的一项研究表明,乳酸可以通过在组蛋白赖氨酸(K)残基上添加一个乳酸基来诱导乳酸化过程,从而调节基因转录。用抗Snail1抗体免疫沉淀,再用抗泛乙酰赖氨酸(Kac)或抗泛乙酰赖氨酸(Kla)抗体免疫印迹显示,缺氧诱导Snail1乙酰化和乳酸化。乳酸进一步上调Snail1乙酰化和乳酸化(图8D)。先前的研究发现,Snail能够与CREB(腺苷3 ',5 ' -单磷酸反应元件结合蛋白)-结合蛋白(CBP)/p300相互作用,诱导其乙酰化。作者观察到Snail1和CBP/p300之间存在相互作用,并且乳酸处理促进了它们在缺氧后的相互作用(图8D)。然而,CHC对细胞内乳酸的抑制减弱了乳酸促进的Snail1与CBP/p300之间的相互作用,以及Snail1的乙酰化和乳酸化(图8E)。相反,与缺氧组相比,乳酸处理也显著增加了Snail1在含有抗cbp抗体、抗p300抗体、抗pan- kla抗体和抗pan- kac抗体的免疫沉淀中的表达(图8F)。相反,CHC降低了Snail1与CBP/p300之间的相互作用,以及Snail1的乙酰化和乳酸化(图8F)。
10.Snail1沉默可减弱缺氧后EndoMT和TGF-β/Smad2的激活
此外,作者揭示了乳酸是否通过Snail1激活促进EndoMT和TGF-β/Smad2的激活。
通过转染Snail1的特异性siRNA使其沉默,逆转了乳酸在缺氧后诱导的CD31和VE-cadherin的下调(图9A)。相反,Snail1抑制可阻止乳酸加速间充质标志物α-SMA和FSP1表达(图9A)、SMAD2磷酸化和TGF-β激活(图9H)。作者还进行了qRT-PCR,观察到在乳酸处理下,Snail1的敲低提高了PECAM1、KDR和CDH5的mRNA水平(图9B至D),降低了ACTA2、COL1A1和S100A4的mRNA水平(图9E至G)。此外,沉默Snail1也减弱了乳酸诱导的SMAD2和TGFB1 mRNA水平(图9I和J)。
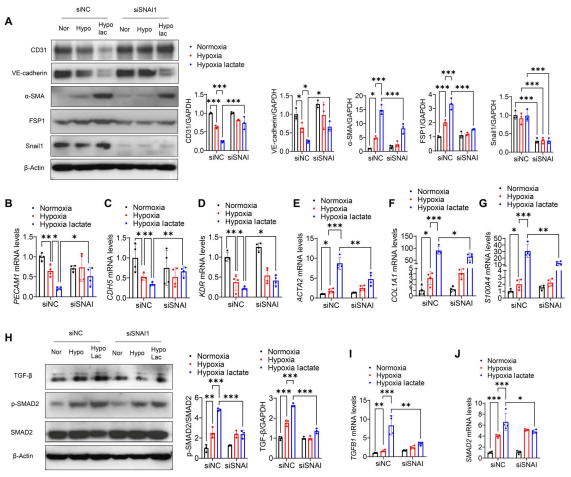
图9. Snail1的沉默会减弱缺氧后EndoMT和TGF-β/Smad2的激活
11.Snail1沉默可改善心肌梗死后的心功能,降低心肌梗死后的EndoMT
为了阐明Snail1在体内是否在EndoMT中发挥作用,作者评估了心肌梗死后Snail1的乳酸化。如图10 (A和B)所示,与假手术组相比,心肌梗死显著促进了Snail1的乳酸化和乙酰化,以及Snail1与CBP/p300之间的相互作用。乳酸给药进一步诱导它们相互作用(图10A)。然而,2-DG处理减弱了MI诱导的Snail1与CBP或p300之间的相互作用,并降低了Snail1的乙酰化和乳酸化(图10B)。然后,作者转染Snail1特异性siRNA,敲低Snail1在心肌中的表达,并研究其是否能改善心功能。免疫印迹和siSnai1 immunofluorescent染色证实,政府会明显降低Snail1表达心中(图10C和D)。手术后7天,
作者观察到击倒Snail1没有对心脏功能的影响,EndoMT虚假的老鼠(无花果。S10, I)。然而,Snail1沉默逆转lactate-decreased EF %和FS %的水平(图10E和F)和阻止LVEDV LVESV乳酸水平引起的心肌梗死后(图10 G和H),这表明缺乏Snail1可以改善乳酸促进的心功能障碍。此外,与心肌梗死后的乳酸处理相比,Snail1沉默提高了Cdh5和Kdr mRNA的表达(图10I和J),降低了Acta2, Fn1和S100a4 mRNA的表达(图10,K至M),这表明Snail1可能有助于心肌梗死后乳酸诱导的EndoMT。总之,作者的数据表明Snail1通过激活TGF-β/Smad2信号在乳酸促进的EndoMT中发挥重要作用(图10N)。
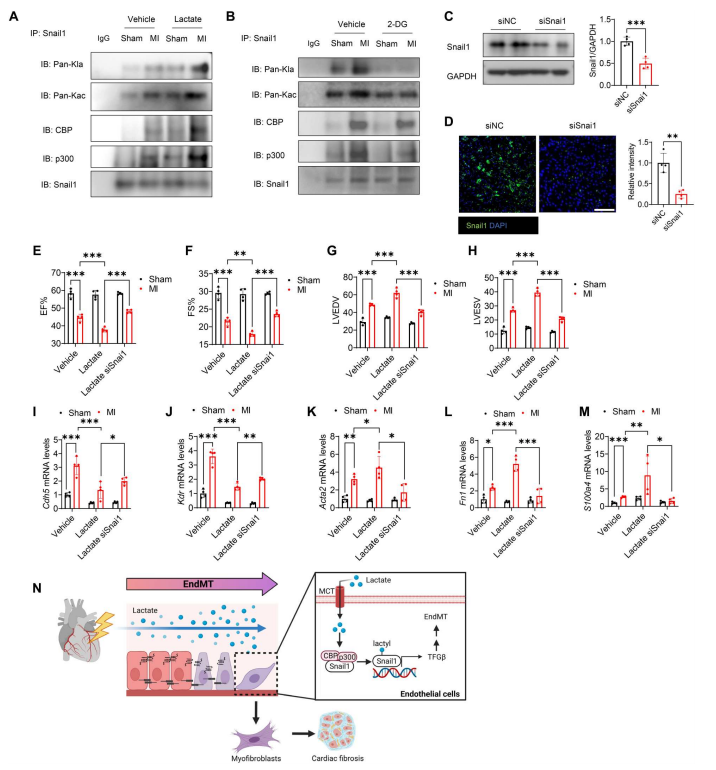
图10. Snail1的沉默会减弱心肌梗死后Snail1的乳酸化和EndoMT
总之,作者的研究揭示了之前未被认识到的乳酸在心肌梗死/缺氧后促进EndoMT的作用。乳酸诱导Snail1的乳酸化和核易位,从而通过激活TGF-β/Smad2信号通路调节EndoMT。这一发现提供了以前未知的见解,即乳酸发挥重要的生物学功能,有助于心肌纤维化的增加和心功能障碍的恶化。
实验方法
心梗模型的诱导、超声心动图、细胞转染、Masson三色染色法、2,3,5-氯化三苯基四氮唑染色、凋亡实验、乳酸测定、流式细胞术、细胞迁移实验、增殖实验、血管生成实验、胶原凝胶收缩试验、免疫荧光染色、WB、RT-PCR、免疫沉淀反应、ChIP-qPCR
参考文献
Fan M, Yang K, Wang X, Chen L, Gill PS, Ha T, Liu L, Lewis NH, Williams DL, Li C. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci Adv. 2023 Feb 3;9(5):eadc9465. doi: 10.1126/sciadv.adc9465. Epub 2023 Feb 3. PMID: 36735787; PMCID: PMC9897666.