






一把铁做的双刃剑——在压力应激中的随机应变
铁是造血和各种关键细胞功能所必需的矿物质。在许多疾病和失调症中,人们越来越多地观察到铁代谢的改变,但对铁代谢受损对细胞的影响仍缺乏全面的机理认识。本文中,作者研究铁过载或缺铁对细胞应激反应和自噬的影响,它们可调控细胞稳态和存活。研究结果显示急性铁负荷导致线粒体ROS(mtROS)的产生和损伤增加、脂质过氧化、自噬通量受损和铁死亡。铁诱导的mROS过度产生是脂质过氧化增加、自噬功能受损和诱导铁死亡的机制。铁过量诱导的铁死亡依赖于细胞类型,并受激活转录因子4(ATF4)的调控。上调ATF4可减轻铁诱导的自噬功能障碍和铁死亡,而沉默ATF4表达则会损害自噬功能,导致mtROS生成增加和铁死亡。利用自噬缺陷的肝细胞和不同的自噬抑制剂,发现自噬损伤使细胞对铁诱导的铁死亡更敏感。相反,缺铁会激活内质网(ER)应激反应,减少自噬并诱导细胞凋亡。缺铁相关的自噬减少是由于ER应激,表现为使用4-PBA降低ER应激反应可改善自噬通量。缺铁导致自噬减少的机制是由于溶酶体膜蛋白翻译后成熟受损导致溶酶体生物发生紊乱。总之,铁过量和缺铁失会导致不同形式的细胞应激和死亡,部分是通过自噬功能受损这一共同机制造成的。本文于2022年7月发表在《Redox Biology》IF:11.8期刊上。
技术路线

主要实验结果:
1、过量的铁诱导氧化反应和自噬的变化但不影响ER应激蛋白
由于细胞对铁含量改变的反应不同,作者确定了铁过量对多种细胞类型的影响,包括人肝癌细胞系HepG2、非转化的小鼠肝细胞系AML12和小鼠巨噬细胞系RAW264.7,以及原代细胞P-Hepa和人主动脉内皮细胞HAECs(图1)。之所以选择这些细胞类型,是因为肝细胞和巨噬细胞在铁的储存和再循环中起着关键作用,血管内皮细胞是铁超载介导损伤的主要靶点之一。
所有的细胞都显示NRF2表达升高以响应FAC铁过载诱导,与HepG2和RAW264.7细胞相比,AML12细胞对NRF2的诱导更为急性和强烈(图1A)。ATF4在AML12和RAW264.7细胞中的诱导作用明显大于HepG2细胞(图1A)。HepG2和AML12细胞中没有诱导包括CHOP、Grp78和P-PERK在内的ER应激标志物,只有RAW264.7细胞中CHOP表达增加,总体上表明过量铁不诱导ER应激(图1A)。HepG2细胞中LC3B-II、活性亚型和p62的表达增加,相反,在AML12和RAW264.7细胞中LC3B的表达无明显改变(图1A)。尽管在AML12细胞中,FTH1在较早的时间点被强烈诱导,但细胞环境依赖性的自噬调节并不是由于基于FTH1诱导的铁负荷的大小(图1A)。在原代培养细胞HAECs和P-Hepa中进一步检测过量铁对NRF2、ATF4和自噬因子表达的影响。两种细胞类型NRF2、p62和LC3B-II的表达均增加,但ATF4的表达未增加(图1B)。与细胞系相比,铁负荷对原代细胞中自噬标志物LC3B-II和p62的影响更强。通过使用FeDex处理WT小鼠,在肝脏(过量铁沉积的主要部位)中测定了急性铁负荷对氧化和自噬标志物表达的体内影响。FeDex注射显著增加FTH1的表达,同样,NRF2、LC3B和p62的表达显著增加,但ATF4的表达没有变化(图1C)。总之,结果表明,急性铁超载会诱导氧化反应和自噬蛋白表达的改变,但不会引起ER应激反应。

图1 过量的铁诱导应激和自噬蛋白的差异表达
2、铁过量引起自噬通量的损害,从而增强铁凋亡
LC3B-II表达升高可提示自噬刺激或自噬通量受损。通过使用自噬抑制剂BafA1或Chlq,确定P-Hepa、HepG2细胞和HAECs中LC3B-II表达的增加是否是自噬通量降低的结果。FAC诱导P-Hepa中p62和LC3B-II的表达在8 h和24 h时间点均显著增加;与BafA1单独处理相比,FAC/BafA1共处理并未进一步增加p62或LC3B-II水平(图2A)。FAC处理诱导P-Hepa中LC3B阳性点状细胞大量聚集;一些铁处理的P-Hepa显示强而弥漫性的LC3B染色(箭头)贯穿整个细胞质以及细胞核的收缩;高倍镜下,这些细胞含有大空泡,完整性完全丧失(图2B),表明细胞死亡。重要的是,铁诱导的LC3B-II积累可被Fer-1阻断(图2C)。这些结果表明,过量的铁诱导脂质过氧化依赖性铁死亡,而清除脂质过氧化物可挽救铁诱导的自噬流抑制。

图2铁过量诱导脂质过氧化依赖性自噬损伤
鉴于过量的铁只在自噬功能障碍的细胞中诱导铁死亡,而自噬在清除氧化分子和受损细胞器方面起着关键作用,所有作者利用P-Hepa 细胞模型想知道自噬功能受损是否是铁过量诱导铁死亡的原因。与单独使用FAC相比,FAC/Chlq共处理导致PI+细胞的百分比增加约3.7倍(图3A)。同样,与单独使用FAC相比,FAC和自噬抑制剂E64d/epstatin A共处理导致PI阳性细胞的百分比增加2.6倍(图3B)。没有利用BafA1来确定铁诱导的细胞死亡,因为它通过提高核内体和溶酶体的pH值来干扰细胞内铁的活动。用4-OHT处理P-Hepa敲除Atg5(ATG5Δhep)(图3C)。与上述自噬通量受损的情况相一致,自噬缺陷也增强FAC诱导的铁死亡,表现为ATG5Δhep细胞中PI阳性细胞的死亡率高于对照细胞(图3D)。这些发现表明,至少在铁过量的情况下,自噬功能障碍会增强铁死亡。
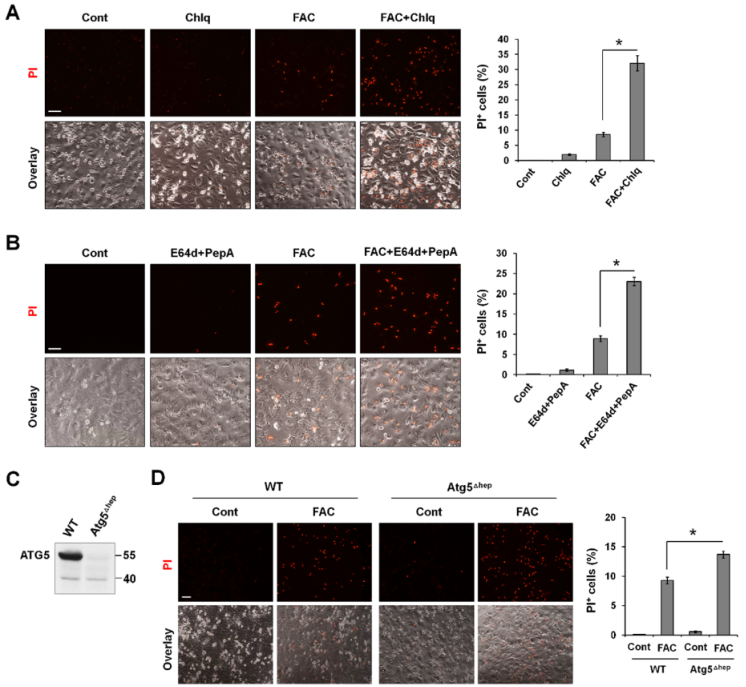
图3 受损的自噬增强铁诱导的铁死亡
3、铁诱导的mtROS产生是脂质过氧化依赖性铁死亡的基础
铁催化的ROS破坏细胞器。TEM成像显示,经FAC处理的P-Hepa细胞的线粒体破裂,嵴(箭头)缺失,这是铁死亡的一个重要特征(图4A)。作者推测铁在线粒体中的负载催化了介导线粒体损伤的mtROS的产生。为探讨这种可能性,用Mito-FerroGreen染色Fe2+,并用MitoSOX检测mtROS。在FAC处理6小时后,观察到线粒体Fe2+和ROS的强烈叠加(图4B)。通过使用荧光探针JC-1检测发现FAC处理导致MMP减少,红色荧光强度明显降低(图4C)。这些发现表明,线粒体中的急性铁负荷会导致mtROS生成过多和MMP 损失,从而导致线粒体损伤。
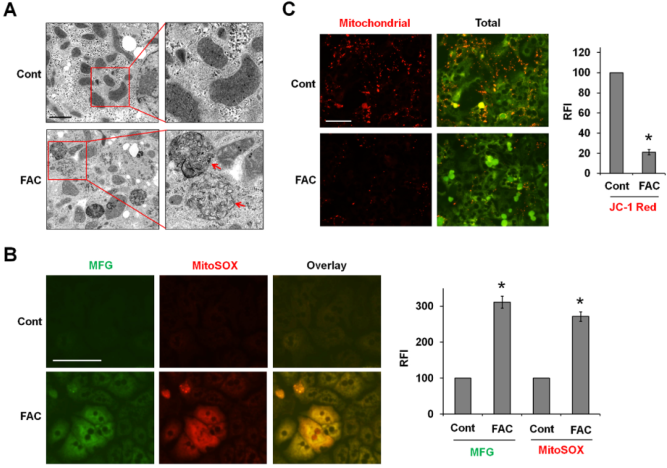
图4铁离子被装载在线粒体中,诱导ROS的产生、MMP的丢失和线粒体损伤
上述研究结果表明,过量产生的线粒体ROS可能是FAC诱导铁死亡的原因。作者研究了常用的线粒体靶向ROS清除剂MitoTEMPO(MT)是否能挽救铁诱导的铁死亡。重要的是,在P-Hepa中,MT和Fer-1都能消除FAC诱导的铁死亡(图5A)。荧光探针BODIPY 581/591在氧化时会变成绿色,通过对其染色,证明MT阻断铁介导的脂质过氧化的效果与铁氧化抑制剂Fer-1不相上下(图5B)。MT还能阻止铁诱导的LC3B-II和p62的积累(图5C),这表明自噬通量得到了改善。这些数据表明,mtROS的过度产生增加了脂质过氧化,这是铁诱导铁变态反应的基本机制。
鱼藤酮(Rotenone)是线粒体复合物I的选择性抑制剂,在高浓度下可诱导mtROS生成并导致细胞凋亡。作者探究低剂量的鱼藤酮与无毒的FAC浓度相结合诱导的mtROS升高是否会使肝细胞对铁依赖性铁死亡敏感。结果显示单独使用鱼藤酮不会诱导PI阳性,而20μM的FAC只导致少量PI阳性细胞(图5D)。与此相反,当FAC和鱼藤酮一起处理P-Hepa时,会导致大量PI 染色,这表明在mtROS产生水平较低的情况下,即使线粒体铁的少量增加也会引发铁死亡。
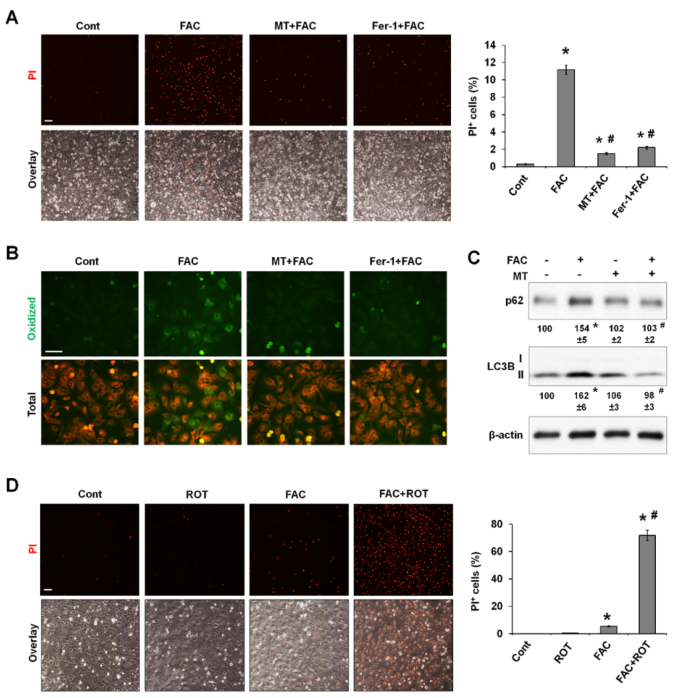
图5 mtROS的过度生成在铁诱导的脂质过氧化、自噬功能障碍和铁死亡中必不可少
4、诱导ATF4保护自噬和细胞存活免受铁毒性
前面研究结果表明,ATF4可维持自噬和细胞存活,抵御铁的毒性。为测试ATF4的潜在作用,利用293 T细胞确定沉默ATF4是否会导致自噬和细胞存活失去对铁毒性的保护。用shATF4质粒构转染293 T细胞,实现ATF4的敲除(图6A)。黄色点相对于总数的百分比增加表明自噬通量受损。在对照细胞中,对照条件下黄色点仅占总点数的7%,用FAC处理也没有引起显著变化(图6B)。与此相反,在对照条件下,ATF4基因敲除会降低自噬通量,黄色点的比例为25%,经FAC处理后显著增加至43%,表明自噬通量进一步降低(图6B)。同样,与对照细胞相比,ATF4缺陷细胞会引起LC3B-II的积累,而FAC会增强LC3B-II 的积累(图6C)。此外,敲除ATF4导致FAC诱导的mtROS生成明显增加(图6D)。与对照细胞相比,shATF4细胞对FAC诱导的细胞死亡敏感,而铁死亡抑制剂Fer-1可减轻这种敏感性(图6E)。总之,结果表明ATF4在维持自噬功能和防止铁诱导的mtROS介导的铁死亡中的作用。

图6 ATF4缺乏导致铁依赖性自噬功能障碍、mtROS产生和铁死亡
5、缺铁诱导内质网应激和自噬减少
为比较铁增加和减少的影响,测定缺铁对不同细胞物种的细胞应激反应和自噬的影响。铁缺乏是由去铁胺(DFO)诱导的,并再次测量FTH1的表达,以显示细胞内的铁储存。之前研究表明,AML12和RAW264.7细胞在自噬和细胞存活变化方面对铁超载不敏感。相反,这两种细胞系都对缺铁敏感,表现出ER应激反应,表现为磷酸化ERK、ATF4和CHOP的表达增加以及LC3B的表达增加(图7A)。这些效应也出现在P-Hepa细胞中,但不出现在HepG2细胞中(图7B)。氧化应激标志物NRF2的表达在长期缺铁的情况下有所下降(图7A)。在AML12细胞中使用自噬抑制剂Chlq,继续评估缺铁如何调控自噬通量。DFO再次诱导LC3B-II表达的显著增加,但与单独使用Chlq相比,DFO/Chlq联合处理并没有进一步增加LC3B-II(图7C),这表明缺铁时自噬通量减少。此外,DFO处理的AML12细胞中裂解的Caspase-3(CC3)表达增加,表明长期缺铁导致细胞凋亡(图7D)。
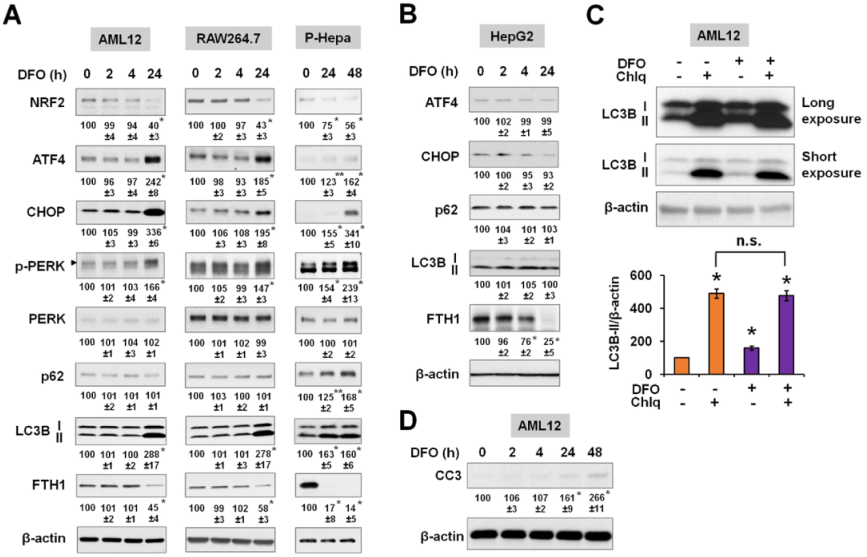
图7铁缺乏可诱导内质网应激反应并降低自噬通量
通过使用肠上皮特异性铁输出蛋白(Fpn)基因敲除小鼠(FpnΔIEC)确定体内缺铁对肝脏ER应激反应和自噬的影响。与之前的报道一致,FpnΔIEC小鼠在Fpn基因敲除6周后出现生长迟缓和明显贫血(脚趾呈灰色)(图8A)表现为FTH1的表达明显降低(图8B)。与对照组Fpnf/f小鼠相比,FpnΔIEC小鼠p-ERK和CHOP的表达明显升高(图8B),表明ER压力增加。缺铁肝脏中CC3的表达也增加(图8B)。同样,荧光染色显示FpnΔIEC小鼠肝脏中Grp78和CC3的表达增加(图8C)。与DFO诱导的培养细胞急性缺铁不同,长期缺铁的FpnΔIEC肝脏中p62和LC3B蛋白表达量显著下降(图8B)。文献表明ER应激可诱导转录重编程,导致许多基因的转录抑制。事实上,FpnΔIEC肝脏中的LC3和p62转录本明显减少(图8D),这也是LC3B和p62蛋白减少的原因。此外,Chlq使对照组肝脏中的 LC3B-II 表达量明显增加了65%,但在FpnΔIEC肝脏中却没有引起任何明显变化(图8E)。同样,Chlq可使对照组肝脏中p62的表达增加90%,但在FpnΔIEC肝脏中仅增加43%。LC3B-II和p62在Chlq作用下的积累减少表明缺铁肝脏的自噬通量下降。
缺铁诱导的ER应激可通过TEM分析再现,在FpnΔIEC肝细胞中可看到轻微扩张的ER(图8F)。在FpnΔIEC 肝细胞中看到糖原(红色箭头)和脂滴(蓝色箭头)堆积增加(图8F)。与之前的报告一致,缺铁的FpnΔIEC肝细胞中线粒体明显增大(图8G),引人注目的是,与野生型肝细胞相比,FpnΔIEC 肝细胞中溶酶体的数量明显减少(图8H),这至少是自噬通量减少的部分原因。
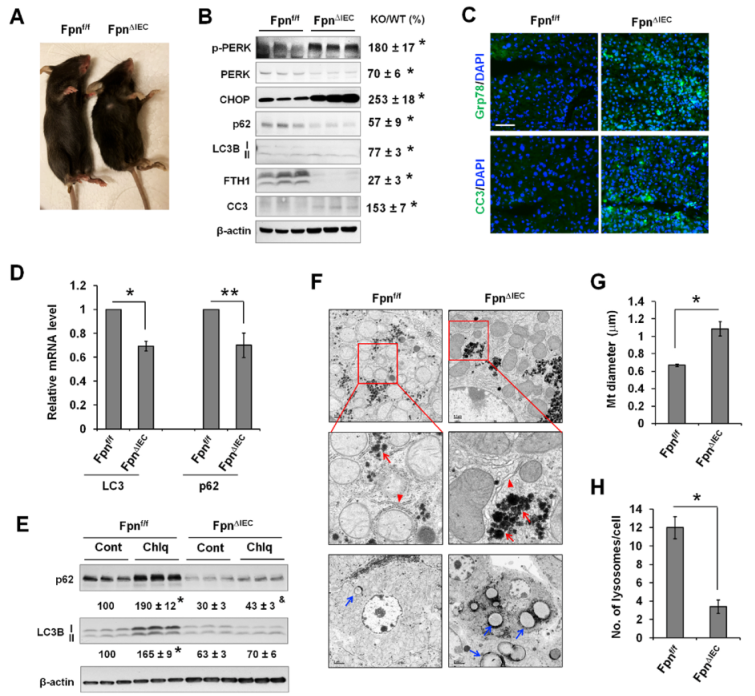
图8铁缺乏诱导内质网应激,并通过减少体内肝脏溶酶体生物发生而降低自噬
6、铁缺乏引起的自噬通量降低依赖于ER应激的诱导
作者试图确定与缺铁相关的ER平衡紊乱是如何导致自噬通量下降的。LAMP1是一种糖基化蛋白质,是溶酶体生物生成和自噬不可或缺的组成部分。结果显示ER应激的DFO和曲卡霉素,两种通过抑制蛋白N-糖基化的自噬诱导剂,都降低LAMP1的成熟(图9A)。4-PBA能帮助蛋白质折叠并抑制ER应激,它能抑制DFO诱导的Grp78表达并改善LAMP1的成熟(图9B)。重要的是,4-PBA可减轻DFO诱导的LC3B-II的积累。
作者推测溶酶体蛋白的成熟缺陷会损害溶酶体的生物生成。对溶酶体蛋白酶 Cathepsin B进行染色[50],以检测溶酶体数量的潜在变化。LysoTracker没有被使用,因为它既能标记溶酶体,也能标记晚期核内体。DFO处理导致cathepsin B 阳性点的数量明显减少,而与4-PBA联合处理可部分缓解这一现象(图9C)。通过使用自噬通量报告基因GFP-LC3-RFP,进一步发现,DFO诱导的黄色点状突起的百分比显著增加,而与4-PBA共同处理后,黄色点状突起的百分比显著减少(图9D),这表明蛋白质成熟的恢复和ER应激的抑制改善了自噬通量。值得注意的是,与未处理的对照组相比,4-PBA 本身也会导致黄色点的百分比轻微但显著地增加。总之,结果表明,缺铁供应扰乱了ER平衡和蛋白质成熟,至少是溶酶体生物生成受损和自噬减少的部分原因。
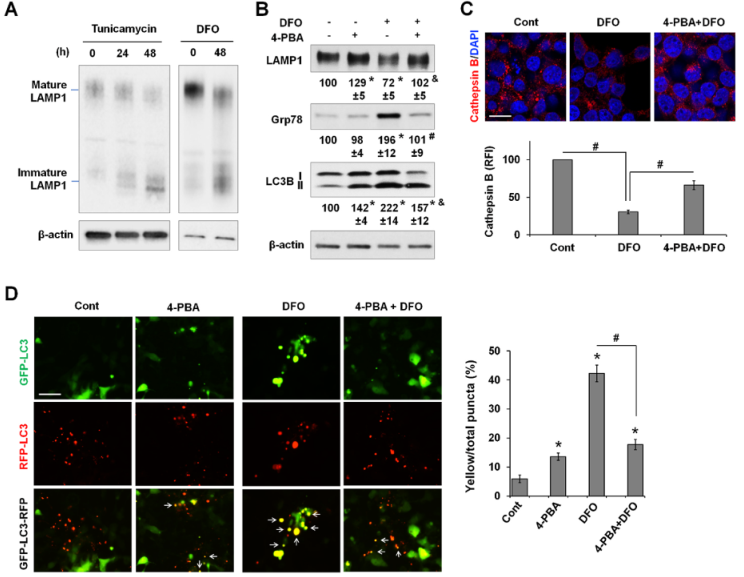
图9缺铁导致的自噬减少与内质网应激有关
总之,本研究表明,一方面铁过量会诱导产生mtROS,从而引起脂质过氧化反应依赖性铁死亡和自噬通量抑制。自噬的减少进一步加强了铁过量诱导的铁死亡。ATF4的上调以及使用mtROS清除剂MT或过氧化脂质清除剂Fer-1可阻碍脂质过氧化和自噬通量的受损,从而阻止铁过量诱导的铁死亡。另一方面,缺铁会损害蛋白质折叠和成熟,导致ER应激和溶酶体生物生成减少,共同导致自噬通量下降。4-PBA可改善蛋白质的成熟并减轻ER压力,从而挽救溶酶体的生物生成和自噬通量。推测ER应激和自噬通量受损都有助于诱导细胞凋亡。
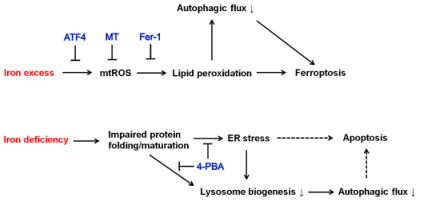
图10 铁过载和铁缺乏对细胞压力响应,自噬,细胞存活影响的示意图
实验方法
qRT-PCR,WB,PI染色检测细胞死亡,共聚焦免疫荧光,ROS,Fe2+,线粒体膜电位,脂质过氧化实验,透射电子显微镜,自噬通量
参考文献
Wang Y, Wang M, Liu Y, Tao H, Banerjee S, Srinivasan S, Nemeth E, Czaja MJ, He P. Integrated regulation of stress responses, autophagy and survival by altered intracellular iron stores. Redox Biol. 2022 Jul 14;55:102407. doi: 10.1016/j.redox.2022.102407.