






抑制“行走的杀手”骨关节炎的“利器”之抑制PIM-1激酶
骨关节炎(OA)是一种炎症性关节炎,巨噬细胞驱动的滑膜炎被认为与软骨破坏密切相关,可发生在任何阶段。然而,目前还没有有效的治疗骨性关节炎进展的靶点。滑膜巨噬细胞中的NOD-、LRR-和pyrin结构域蛋白3(NLRP3)炎症小体参与病理性炎症过程,所以针对它的治疗策略被认为是治疗OA的有效途径。PIM-1激酶作为多种细胞因子信号通路的下游效应者,在炎症性疾病中发挥促炎作用。
在这项研究中,研究者评估了PIM-1的表达和滑膜巨噬细胞在人骨性关节炎滑膜中的渗透。以小鼠和人巨噬细胞为模型,研究了PIM-1对脂多糖(LPS)和尼日利亚菌素、三磷酸腺苷、尿酸钠(MSU)、铝盐等激动剂的作用及其机制。采用改良的巨噬细胞条件培养液(CM)共培养体系评价其对软骨细胞的保护作用。体内疗效通过内侧半月板(DMM)诱导的小鼠骨性关节炎得到证实。
PIM-1在人骨性关节炎滑膜中的表达增加,并伴有滑膜巨噬细胞的浸润。在体外实验中,特异性抑制剂SMI-4a对PIM-1的抑制作用迅速抑制了小鼠和人巨噬细胞中NLRP3炎症小体的激活以及gasdermin-D(GSDME)介导的细胞焦亡。此外,PIM-1的抑制在组装阶段特异性阻断了含有CARD(ASC)寡聚化的凋亡相关斑点样蛋白。机制上,PIM-1的抑制可减轻线粒体活性氧(ROS)/氯离子胞内通道蛋白(CLICs)依赖的Cl -外泄信号通路,最终导致ASC寡聚化和NLRP3炎性体活化受阻。此外,PIM-1的抑制在改良的共培养系统中显示出软骨保护作用。最后,SMI-4a显著抑制滑膜中PIM-1的表达,并降低DMM诱导的OA模型中的滑膜炎评分和国际骨关节炎研究协会(OARSI)评分。本文于2023年7月发表在《Journal of Translational Medicine》期刊上,IF=7.4。
技术路线

主要实验结果
1、人骨关节炎滑膜巨噬细胞PIM-1表达上调
探讨PIM-1在巨噬细胞和骨性关节炎进展中的作用。研究者研究了巨噬细胞标志物(F4/80)在人滑膜中的表达。与之前的研究一样,与正常滑膜相比,骨性关节炎患者滑膜中F4/80阳性细胞的数量增加(图1a,b)。此外,在骨性关节炎患者的滑膜中,PIM-1阳性细胞的百分比也升高(图1a,c)。有趣的是,PIM-1主要定位于巨噬细胞,PIM-1和F4/80共存。综上所述,这些结果表明巨噬细胞在OA滑膜中聚集,并增加PIM-1的表达。

图1 巨噬细胞和PIM-1在OA滑膜中表达上调
2、PIM‑1的抑制抑制了NLRP3炎性体的激活
为了确定Smi-4a是否特异性地抑制巨噬细胞中的PIM-1,研究者用Smi-4a对脂多糖诱导的BMDM(骨髓源性巨噬细胞)进行了2 h的蛋白质印迹分析。结果表明,SMI-4在LPS刺激(3.3 μm, 10 μm, 30 μm)后可下调jak2、stat的磷酸化水平,并呈剂量依赖性(图2a)。这意味着PIM-1活性被SMI-4a抑制。此外,研究者在尼日利亚菌素或ATP刺激用Smi-4a培养脂多糖诱导的BMDM。结果表明,SMI-4a以剂量依赖性(3.3 μm, 10 μm, 30 μm抑制pro-IL-1β(P31)和pro-caspase-1(P45)裂解为IL-1β(P17)和caspase-1(P20)(图2b、c),但不影响TNF-a的分泌。生长因子IL-3可通过JAK/ STAT信号通路上调PIM-1的活性。值得注意的是,研究者的研究结果表明,添加IL-3(25和50ng/ml)可以恢复SMI-4a(10 μm)诱导的jak2、stat的低磷酸化水平(图2d)。这意味着补充IL-3(25和50ng/ml)可以恢复PIM-1的活性。此外,PIM-1的激活能够“挽救”SMI-4a(30 μm)诱导的IL-1β分泌下调(图2e和f),而不改变TNF-a分泌。这些发现表明,PIM-1可以参与NLRP3炎性小体激活的组装阶段,而不影响启动阶段。
为了观察PIM-1对巨噬细胞中NLRP3炎症小体的启动阶段和组装阶段的作用,在LPS刺激前和刺激后应用SMI-4a。结果表明,在LPS刺激前预处理SMI-4a抑制了TNF-α的产生、pro-IL-1β和NLRP3蛋白的表达,而在LPS刺激后用SMI-4a处理时,这些没有明显变化(图2g–i)。这些发现表明,PIM-1的抑制可抑制NF-κB信号通路,并可在启动期和装配期抑制NLRP3炎症小体的激活。研究者继续探索当巨噬细胞在LPS刺激后用SMI-4a处理时,PIM-1在装配阶段的作用。有趣的是,被SMI-4a抑制的PIM-1显示出对NLRP3炎症小体激活的快速抑制作用(图2j–l)。为了进一步消除SMI-4a脱靶的可能性,这项研究还发现,另一种PIM-1抑制剂AZD1208可以快速抑制NLRP3炎症小体激活的组装阶段,而不影响启动阶段。
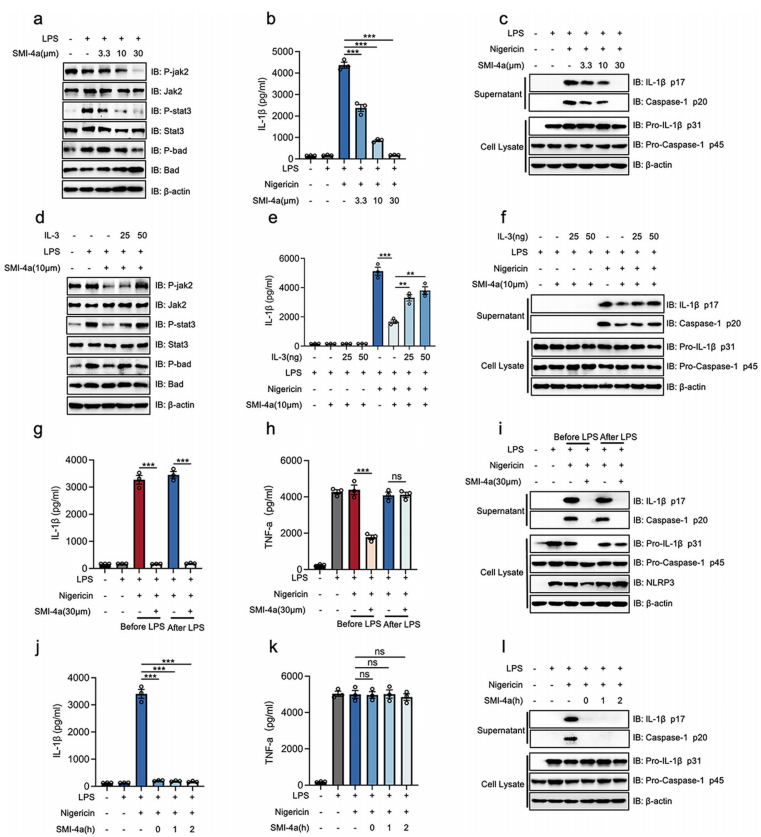
图2 . PIM-1的阻断抑制了巨噬细胞NLRP3炎性体活化
3、阻断PIM‑1可广泛抑制NLRP3、AIM2、NLRC4炎性体和焦亡,并特异性减轻ASC低聚物的形成
为了进一步研究PIM-1是否在NLRP3炎性小体激活中起共同作用,研究者在不同的巨噬细胞中使用不同的激动剂研究了PIM-1对NLRP3炎性小体激活的作用。研究者发现SMI-4a剂量依赖性地抑制小鼠和人巨噬细胞NLRP3炎性体的激活,如雄性C57BL/6小鼠的PMs(图3a, b),THP-1细胞(图3c, d),健康人的外周血单核细胞(PBMC)。除尼日利亚菌素外,SMI-4a还能抑制MSU和Alum诱导的caspase-1裂解和IL-1β分泌(图3e, f)。
为了探究PIM-1是否在NLRP3炎性小体激活中起特定作用,研究者研究了PIM-1在NLRC4和AIM2炎性小体激活中的作用。结果显示SMI-4a对AIM2炎性小体和NLRC4炎性小体具有抑制作用,可由poly(dA:dT)转染或沙门氏菌感染触发(图3g, h),导致IL-1β产生减少。
LDH释放和GSDMD裂解是炎性小体诱导的焦亡的两个关键特征。然后,研究者发现SMI-4a对PIM-1的抑制可以抑制LDH的释放,阻断GSDMD的裂解(图3i, j)。接下来,研究者研究了PIM-1如何影响NLRP3炎性体形成的组装阶段。NLRP3和ASC之间的相互作用对于NLRP3炎性体的组装至关重要。共免疫沉淀(CO-IP)结果表明SMI-4a不影响NLRP3和ASC的相互作用。在NLRP3和ASC装配后,随后的ASC募集形成ASC寡聚体和ASC斑点。结果首先表明,PIM-1抑制可以特异性减弱尼日利亚菌素诱导的DSS交联的ASC寡聚体和ASC-斑点(图3k - m)。
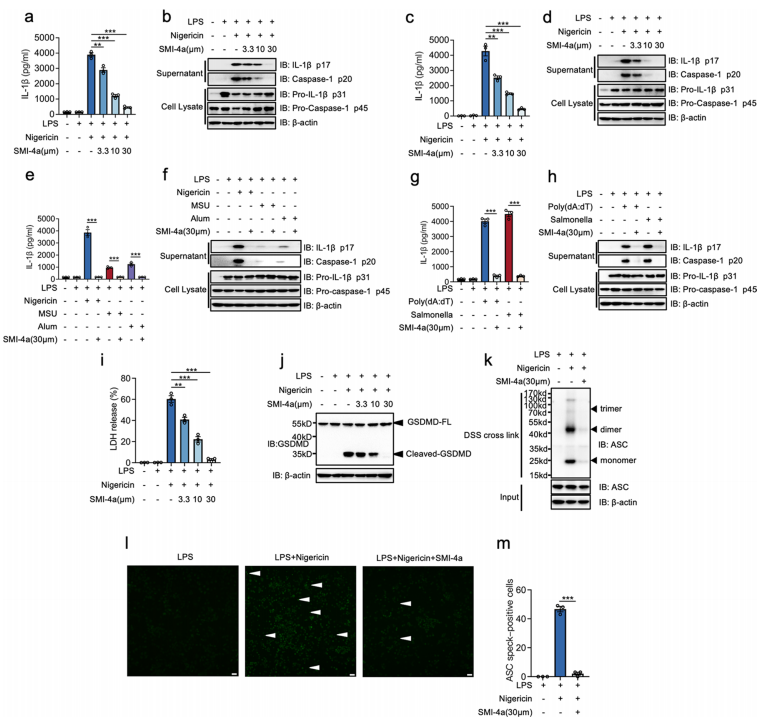
图3 PIM-1的抑制广泛抑制了NLRP3、AIM2、NLRC4炎性体和焦亡,并特异性减轻了ASC低聚物形成
4、PIM-1的抑制阻断了Cl-外排以抑制ASC寡聚化
Cl-外排在ASC寡聚化中起着至关重要的作用,并且是NLRP3炎症小体激活的重要上游事件。为了了解PIM-1对Cl-流出的影响,研究者进行了离子置换实验来驱动K+或Cl-的流出。与以前的研究一致,单独的无氯条件不会引起IL-1β的释放,但它可以进一步增强无钾条件引起的IL-1β的释放。此外,在用SMI-4a处理的这些上清液中没有观察到IL-1β(图4a)。此外,在无K+和Cl-的条件下,SMI-4a可以剂量依赖性地抑制IL-1β的释放(图4b,c)。与先前的研究一致,研究者发现无K+和无Cl-的条件可以激活ASC寡聚化。SMI-4a可以减轻由尼日利亚菌素或无K+和Cl-条件引起的ASC寡聚化(图4d)。以上结果提示,PIM1抑制NLRP3炎性小体激活可能与Cl−外流有关。
如图4e,f所示,nigericin或无K+和Cl-条件显著增加了细胞内荧光强度,表明细胞内Cl-水平降低,Cl-流出增强。但是,这些进程被SMI-4a阻止了。氯离子细胞内通道蛋白(CLICs)家族由CLIC1、CLIC4和CLIC5组成,存在于胞浆和质膜中。CLICs可以移位到质膜上,形成阴离子通道,介导细胞内的Cl-外流。研究者进一步探索SMI-4a诱导的Cl-外排变化是否是通过阻断CLICs转运介导的。蛋白质印迹分析表明,SMI-4a可以阻断由尼日利亚菌素和无K+和Cl-条件诱导的CLICs易位(图4g,h)。综上所述,研究者首次表明PIM-1在CLICs转运和Cl-外流中发挥关键作用,以实现ASC寡聚化和NLRP3炎症小体激活。

图4 PIM-1的抑制可阻断Cl−外排,从而抑制巨噬细胞中ASC的低聚化
5、PIM-1的抑制通过线粒体ROS阻断Cl-外排
线粒体ROS在Cl-流出的上游起作用,加速NLRP3炎症体的激活,并在AIM2和NLRC4炎症体的激活中起关键作用。为了研究PIM-1抑制是否通过调节线粒体ROS阻断Cl-外排和NLRP3炎症小体激活,研究者首先使用MitoSOX红色探针检测巨噬细胞中线粒体ROS的水平。结果显示,伴随线粒体ROS和SMI-4a而产生的NLRP3炎症激活可以抑制线粒体的ROS水平(图5a,b)。此外,在SMI-4a和尼日利亚菌素孵育之前,用鱼藤酮(一种线粒体电子传递链复合物I抑制剂)处理LPS诱导的BMDMs时,线粒体ROS的产生显著增加。这一发现表明鱼藤酮阻断了SMI-4a对线粒体ROS产生的抑制功能(图5c,d)。此外,鱼藤酮预处理后,Cl-外排和NLRP3炎症小体的激活增强(图5e - h)。总之,这些结果首次表明PIM-1的抑制抑制了线粒体ROS的产生并阻断了Cl-外排,导致了对NLRP3炎症小体的抑制作用。
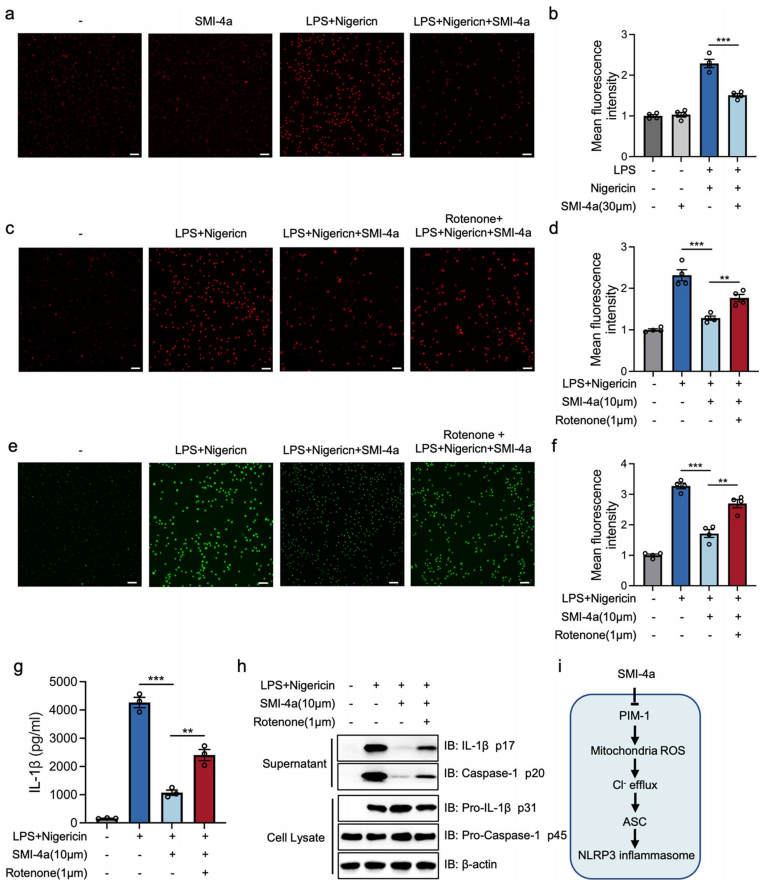
图5 PIM-1的抑制抑制了线粒体ROS的产生,从而阻断了巨噬细胞中Cl-的流出
6、在共培养体系中,PIM-1的抑制抑制了软骨细胞凋亡并改善软骨细胞增殖和ECM代谢
研究者进一步利用共培养系统来研究巨噬细胞和软骨细胞的相互作用。首先,研究者应该检测SMI-4a对软骨细胞的细胞毒性作用。结果显示浓度< 50 µM的SMI-4a 处理软骨细胞24或48小时没有显著的细胞毒性作用(图6b)。随后,研究者用参与NLPR3炎症体激活的各种单一成分刺激巨噬细胞,并用CM培养软骨细胞。有趣的是,与对照组相比,Nigericin组软骨细胞的活性降低,而其他组无明显变化。这意味着从用尼日利亚菌素处理的巨噬细胞收集的CM对软骨细胞具有明显的毒性作用(图6c)。最终,研究者放弃了尼日利亚菌素,选择了无K+和Cl-的条件来激活NLRP3炎症小体。因此,建立了改良的巨噬细胞-软骨细胞共培养系统(图6a)。
在该共培养系统中,与无LPS+KCl组相比,在SMI4a处理下软骨细胞的存活力增加(图6d)。从TUNEL染色中观察到一致的结果(图6g,h)。此外,与无LPS+KCl组相比,SMI-4a处理后,裂解的caspase 3和Bax的表达降低,Bcl-2的表达升高(图6e)。这些结果表明,PIM-1的抑制可以减轻软骨细胞凋亡。此外,与无LPS+KCl组相比,在SMI-4a处理下,胶原II和聚集蛋白聚糖的表达水平增加,MMP13和ADAMTS5的表达水平降低(图6f)。这些结果表明,抑制PIM-1可以恢复软骨细胞的细胞外基质代谢。EDU染色显示,与无LPS+ KCl组相比,用SMI-4a处理的软骨细胞的增殖水平增加(图6i和j)。基于该结果,如Toloniumchloride和S&F染色的染色强度和细胞面积分数的增加所示(图6k – n),用SMI-4a处理的软骨细胞的数量增加。这些结果首次证明PIM-1的抑制对软骨细胞增殖有有益的影响。

图6 在共培养体系中PIM-1的抑制防止了软骨细胞损伤
7、PIM-1的抑制可以改善DMM模型小鼠骨关节炎的进展
该研究进一步探索了PIM-1抑制对体内OA的潜在治疗作用。H&E染色显示,smim4a治疗DMM小鼠改善了滑膜的肥大和增生,并降低了滑膜衬里细胞层的厚度(图7b)。结果表明SMI-4a可以降低滑膜炎评分,该评分在DMM组中升高(图7c)。此外,与人类OA滑膜一致,与巨噬细胞共定位的PIM-1(F4/80)在DMM小鼠中显著上调(图7d–f)。然而,用SMI-4a处理对PIM-1的抑制表明滑膜中的这些阳性细胞显著下调。此外,S&F染色显示,与DMM组相比,SMI-4a的治疗改善了粗糙和糜烂的关节表面软骨、软骨细胞减少和异常狭窄的关节间隙。结果表明SMI-4a可以降低在DMM组中升高的OARSI评分(图7h)。这些蛋白质的免疫组织化学在OA的发展中起着特征性的病理学指示作用,如胶原II、聚集蛋白聚糖、MMP13、ADAMTS5和cleaved-caspase3。与DMM组相比,用SMI-4a处理改善了II型胶原和聚集蛋白聚糖的下调,并抑制了MMP13、ADAMTS5和cleaved-caspase3的上升(图7i)。出于SMI-4a的安全性考虑,测试了肝酶丙氨酸转氨酶(ALT)和天冬氨酸转氨酶(AST)的血清水平的肝毒性(图7j),评估了血尿素氮(BUN)和肌酐(Cre)的肾毒性(图7k),评估了体重的全身毒性(图7l)。与对照组和DMM组相比,这些参数在SMI-4a处理后均无明显变化。此外,从肝、脾和肾的H&E染色中没有观察到明显的变化(图7m)。研究者的体内研究结果首次表明PIM-1抑制可以在OA小鼠中发挥保护作用。
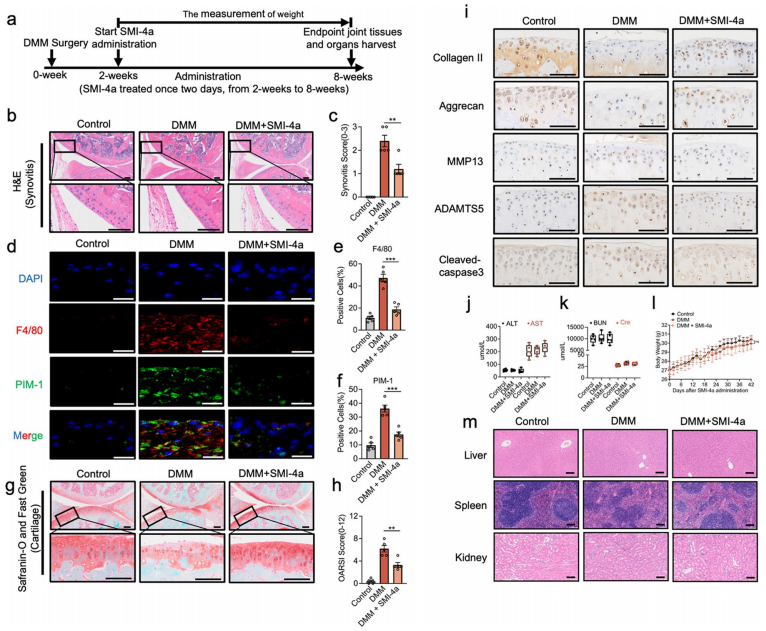
图7 PIM-1抑制改善了小鼠DMM模型中OA的进展
总之,我们的研究强调PIM-1是治疗OA的有效靶点。抑制PIM-1可抑制NLRP3炎症体和GSDMED介导的细胞焦亡,其机制可能是通过阻断组装阶段线粒体ROS/Cl−外流来实现。我们的研究表明,PIM-1特异性抑制剂能有效地治疗小鼠骨性关节炎。因此,PIM-1代表了一类新的有希望的治疗OA的靶点,靶向于巨噬细胞中的这些机制,并拓宽了治疗OA策略的道路。
实验方法
酶联免疫吸附、乳酸脱氢酶释放试验、WB、细胞内氯离子水平的评估、H&E染色、S&F染色
参考文献
Zhang, Z. et al. Targeting macrophagic PIM-1 alleviates osteoarthritis by inhibiting NLRP3 inflammasome activation via suppressing mitochondrial ROS/Cl− efflux signaling pathway. Journal of Translational Medicine 2023 Jul, 21:452 doi:10.1186/s12967-023-04313-1