






代谢重编程角度揭示自噬与糖酵解之间的相互作用的机制

自噬和糖酵解是涉及生理和病理细胞程序的高度保守的生物过程,但这些过程之间的相互作用并不明确。本研究发现了ULK 1直接与LDHA相互作用促进乳酸产生,乳酸通过Vps 34乳酰化连接自噬和糖酵解,Vps 34乳酰化增强了Vps 34与Beclin 1、Atg 14 L和UVRAG的结合,然后增加了Vps 34脂质激酶活性,从而促进了自噬通量和内溶酶体运输。本研究描述了自噬调控机制,整合了两个高度保守的生命过程(糖酵解和自噬)。本文于2023年6月发表于《Science Advances》上,IF=13.6。
技术路线















研究内容
1.ULK1诱导LDHA Ser196磷酸化
ULK 1是参与自噬早期到晚期阶段的最关键的蛋白激酶。为了全面鉴定ULK 1相互作用蛋白,进行了串联亲和纯化。免疫沉淀和酵母双杂交测定证实ULKl与LDHA相互作用(图1A-C)。由于ULK1介导糖酵解酶HK和PFK1的磷酸化,我们推测ULK1可能是一种LDHA激酶。为了检验这一假设,进行了体外激酶测定和质谱分析,发现LDHA在S196被ULK1磷酸化。为了验证LDHA S196的磷酸化,产生特异性识别S196-磷酸化LDHA的多克隆抗体(图1D)。用丙氨酸替换196位的丝氨酸(S196 A)完全消除了LDHA磷酸化(图1E)。此外,通过Earle平衡盐溶液(EBSS)诱导的饥饿或雷帕霉素(mTOR)抑制剂雷帕霉素的机制靶标的ULKl活化增强LDHA S196磷酸化,并且通过ULKl抑制剂MRT68921的ULKl抑制降低LDHA S196磷酸化(图1F)。同样地,与对照细胞相比,ULK1敲除(KO)小鼠胚胎成纤维细胞(MEF)中的LDHA S196磷酸化降低,并且在正常和EBSS培养基中ULK1重新表达后,LDHA S196磷酸化重新建立(图1G)。LDHA敲低抑制p62降解,并且LDHAWT和LDHAS196D的转染完全逆转了LDHA敲低的作用,而LDHAS196A部分挽救了这些作用(图1H)。体外激酶测定和Western印迹进一步证实了LDHAWT的Ser196被ULK1磷酸化,但LDHA突变体(LDHAS96A)在体外不被ULK1磷酸化(图1I)。因此,这些结果表明ULK1作为上游调节因子介导LDHA S196磷酸化。

图1:1.ULK1诱导LDHA Ser196磷酸化
2.LDHA磷酸化增强其酶活性
为了测试磷酸化对LDHA酶活性的生物学效应,通过EBSS培养基或mTOR抑制剂雷帕霉素的ULK 1活化增加了细胞溶质乳酸盐水平,而ULK1抑制剂MRT 68921降低了细胞溶质乳酸盐水平(图2A)。同样地,通过短发夹RNA(shRNA)敲低ULKl表达也降低了正常培养基或EBSS培养基中的细胞溶质乳酸水平,但沉默其他自噬基因(即Vps 34、Atg 5和Atg 7)的表达对细胞溶质乳酸水平没有影响(图2B和C)。此外,ULK 1敲低H1299细胞中ULK 1 WT表达的恢复重新建立了细胞溶质乳酸盐水平,而ULK1 ΔKI突变体(具有丢失的激酶结构域)的表达未恢复(图2D)。氨基酸饥饿和mTOR抑制剂Torinl处理降低了细胞溶质NADH/NAD+比率,然后氨基酸补充和去除Torinl恢复了NADH/NAD+比率(图2 E)。此外,LDHA沉默降低了细胞溶质乳酸水平,并且LDHAWT和LDHAS196D表达的恢复恢复了细胞溶质乳酸水平,而LDHAS196A表达未能挽救LDHA酶活性(图F和G)。这些表型通过体外测量酶活性和体内进行流式细胞术来证实(图2 H)。此外,通过草氨酸盐或shRNA抑制LDHA酶活性和通过MRT 68921抑制ULK1活性降低了在EBSS培养基中培养的细胞中的胞质乳酸盐水平(图2 I)。这些结果表明ULK1介导LDHA磷酸化并增强其酶活性。

图2:LDHA磷酸化增强其酶活性
3.Vps34通过乙酰转移酶TIP60在K356和K781处被乳酰化
为了验证乳酰化在自噬调节中的作用,测量了哺乳动物自噬核心蛋白的乳糖化的免疫共沉淀和蛋白质印迹与抗泛-乳糖化抗体。发现许多在不同自噬阶段的乳酰化自噬核心蛋白(Vps 34、ULK 1、UVRAG等)在细胞内表达(图3A和B)。同时,发现在LDHA敲低细胞中Vps 34乳酰化水平降低(图3C)和LDHA抑制剂草氨酸盐处理的细胞,但乳酸盐、A-7669662(AMPK激活剂)或鱼藤酮处理的细胞中增加(图3D)。为了鉴定Vps 34乳酰化位点,在免疫沉淀后从用IOmM乳酸盐处理24小时的人胚肾(HEK)293 T细胞中纯化Vps 34-Flag,并通过质谱分析,并鉴定了两个赖氨酸残基(K356和K781)。为了验证这两个乳酰化位点,通过定点诱变将两个赖氨酸残基改变为精氨酸残基,并将这些突变体转染到HEK293T细胞中。Vps34K356R和Vps34K781R的乳酰化水平显著降低,并且双突变体Vps342KR的乳酰化水平被消除(图3E)。为了鉴定Vps 34乳酰化书写者,构建乙酰转移酶shRNA文库,并通过免疫共沉淀和用抗泛乳酰化抗体的蛋白质印迹法测定Vps 34乳酰化水平。敲低KAT5/TIP60表达减弱了Vps34的乳酰化,但没有其他乙酰转移酶发挥这种作用(图3F)。为了鉴定与Vps 34相互作用的蛋白质,用二硫代双溶液(DSP)处理过表达Vps 34-Flag的细胞以诱导交联。Vps 34-Flag相互作用蛋白通过免疫共沉淀被拉下,然后通过质谱法鉴定。除了PI3KC3亚基Beclinl、UVRAG、Atgl 4、Pacer和Rubicon之外,Vps 34还与TIP60和p300相互作用(图3G)。内源性和外源性免疫沉淀实验证实了Vps34与TIP60相互作用,并且EBSS饥饿促进了Vps34和TIP60的相互作用(图3H)。通过TIP60抑制剂MG149或TIP60激酶GSK3抑制剂SB216763抑制TIP60活性降低了Vps34乳酰化,并且通过氨基酸或血清剥夺增强TIP60活性增加了Vps34乳酰化水平(图3I)。同样地,用野生型TIP60处理在TIP60敲低的HEK293T细胞中重新建立了Vps34乳酰化的下调,但是功能丧失突变体TIP60S86A未能挽救已经被TIP60敲低的HEK293T细胞中已经降低的Vps34乳酰化(图3J)。体外乳酰化测定显示TIP60通过泛-Kla抗体和通过质谱法使Vps 34 K356和K781乳酰化(图3K)。这些结果表明,TIP 60介导Vps 34乳酰化。
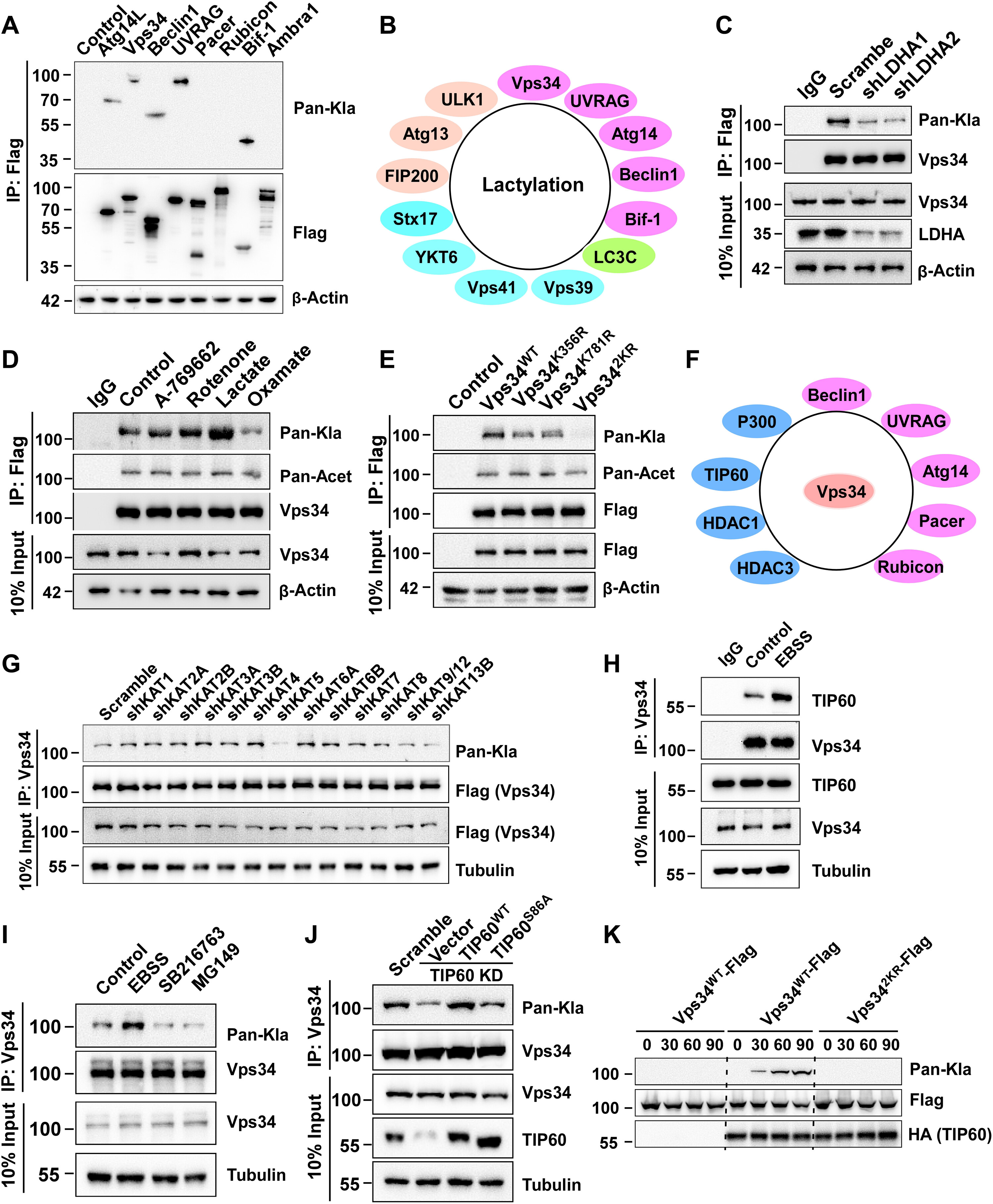
图3:Vps34通过乙酰转移酶TIP60在K356和K781处被乳酰化
4.Vps34乳酰化正调控其与UVRAG和Beclin1的相互作用以及脂质激酶活性
为探讨Vps 34乳酰化的生物学功能,采用免疫沉淀和Western blot分析Vps 34与Vps15、Beclin1、ATG14L和UVRAG的相互作用。通过shRNA介导的LDHA敲低降低Vps 34乳酰化损害了Vps34与Beclinl、ATG14L和UVRAG的结合,并且功能丧失突变体LDHAS196A未能挽救Vps34与LDHA敲低细胞中这些亚基的结合,这与野生型LDHA处理的效果相反(图4A)。Vps 34 K356R和Vps 34 K781R突变体分别使Vps 34-Beclin1-ATG14L复合物(I)和Vps34-Beclin1-UVRAG复合物(II)不稳定,而Vps 342KR突变体在两个乳酰化位点处具有取代,降低了Vps 34与Beclin1、ATG14L和UVRAG在正常培养基和EBSS培养基中的结合(图4B和C)。此外,高乳酸盐处理促进了Vps34WT与Beclin1、ATG14L和UVRAG的结合,而不是Vps342KR的结合(图4D)。此外,与对照细胞相比,当在正常或EBSS培养基中培养的细胞中Vps 34乳酰化写入器KAT5/TIP60被敲低时,Vps34与Beclin1、ATG14L和UVRAG的结合减少(图4 E)。结果表明,Vps 34乳酰化促进了Vps 34与Beclin 1、ATG 14 L和UVRAG的结合。在Vps 34沉默的细胞中转染Vps 34 WT完全恢复了Vps 34敲低的作用,而转染Vps342KR仅部分逆转了这些作用(图4F和G)。同样地,在饥饿条件下,与对照细胞中相比,Vps34沉默减少了GFP-DFCPl斑点的数目,Vps 34沉默的细胞中恢复Vps 34 WT表达完全储存了GFPDFCPl斑点的数目,并且Vps 342 KR表达仅部分逆转了这些效应(图4H)。体内和体外ELISA结果显示Vps 34脱酰化部分损害Vps34脂质激酶活性(图4J和K)。总之,这些结果表明,乳糖化通过增强Vps34与Beclin1、ATG14L和UVRAG的结合来促进Vps 34脂质激酶活性。
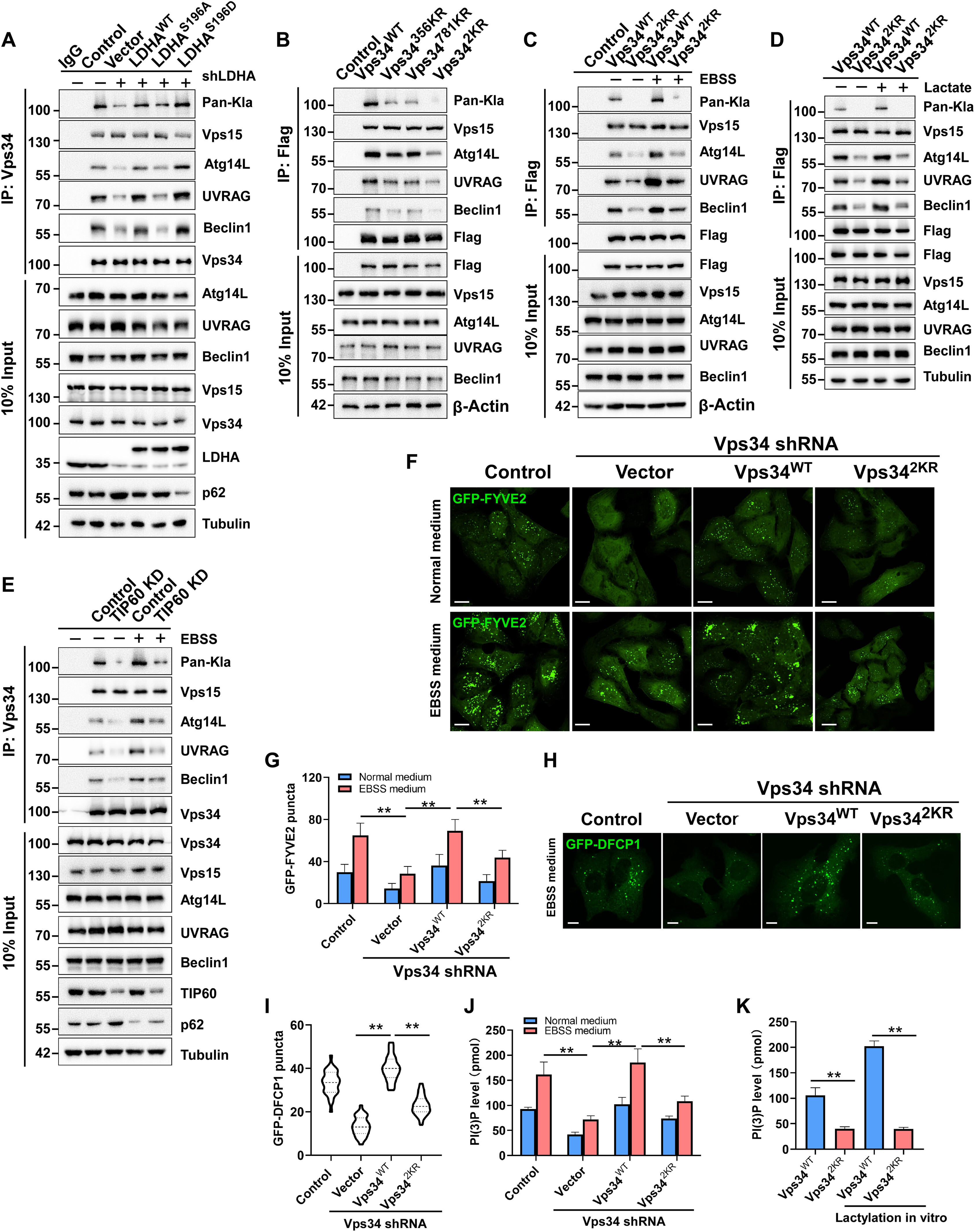
图4:Vps34乳酰化正调控其与UVRAG和Beclin1的相互作用以及脂质激酶活性
5.Vps34乳酰化促进自噬体的形成和成熟
为了分析Vps 34乳酰化对自噬通量的影响,通过蛋白质印迹分析p62降解。Vps 342KR部分地抑制p62降解,与Vps 34 WT在正常培养基中和EBSS饥饿条件下在细胞水平上的作用相反(图5A)。用GFP-LC 3切割测定法验证乳酸和Vps 34乳酰化对自噬通量的生物学功能。Vps 34敲低的HEK293 T细胞中的GFP-LC 3切割在正常培养基和EBSS培养基中受到抑制,并且Vps 34 WT表达的恢复促进了GFP-LC3切割,并且Vps 342KR的再表达仅部分逆转了这些效应(图5 B)。此外,Vps 34敲低的HEK 293T细胞中的GFP-LC 3切割在正常培养基和EBSS培养基中受到抑制,并且Vps 34 WT表达的恢复促进了GFP-LC 3切割,并且Vps 342KR的再表达仅部分逆转了这些效应(图5 B)。这些结果通过内源性LC 3斑点测定进一步证实(图5C和D)。此外,透射电子显微镜测定显示,与正常培养基和EBSS诱导的饥饿条件下培养的对照细胞相比,Vps 34敲低的H1299细胞中自噬囊泡的数量减少,Vps34 WT表达逆转了自噬囊泡的数量减少,而Vps 342KR部分恢复了囊泡的数量(图5E和F)。这些结果表明,Vps 34乳酰化是自噬体形成和成熟所必需的。

图5:5.Vps34乳酰化促进自噬体的形成和成熟
6.Vps 34乳酰化促进内体-溶酶体降解
为了确定Vps34乳酰化是否调节这种表型,在Vps34敲低的U2OS中表达了Vps34WT和Vps342KR。免疫荧光显微镜显示用Vps34WT重建校正了扩大的内体积累的表型,并且Vps342KR部分挽救了内体积累(图6A)。通过Western印迹分析分析乳酸盐介导的Vps34乳酸化是否调节EGFR降解、内源性EGFR降解。在Vps34敲低细胞中转染Vps34WT完全促进EGFR降解,而Vps342KR转染仅部分恢复EGFR降解(图6B)。晚期核内体标志物Rab7和LysoTracker red的共定位显示Vps34敲低阻断了晚期核内体和预先存在的溶酶体的融合,Vps34WT的再表达挽救了这种表型而不是Vps342KR(图6E和F)。由于不充分的内溶酶体融合,我发现Lampl表达通过免疫荧光染色和Western印迹在Vps34敲低的U2OS中增加,但Vps34WT恢复了这种表型,并且Vps34脱酰化突变体失去了这种功能(图6G和H)。这些结果表明,Vps34乳糖化位点的突变体损害了其生物学功能,导致溶酶体降解不足。
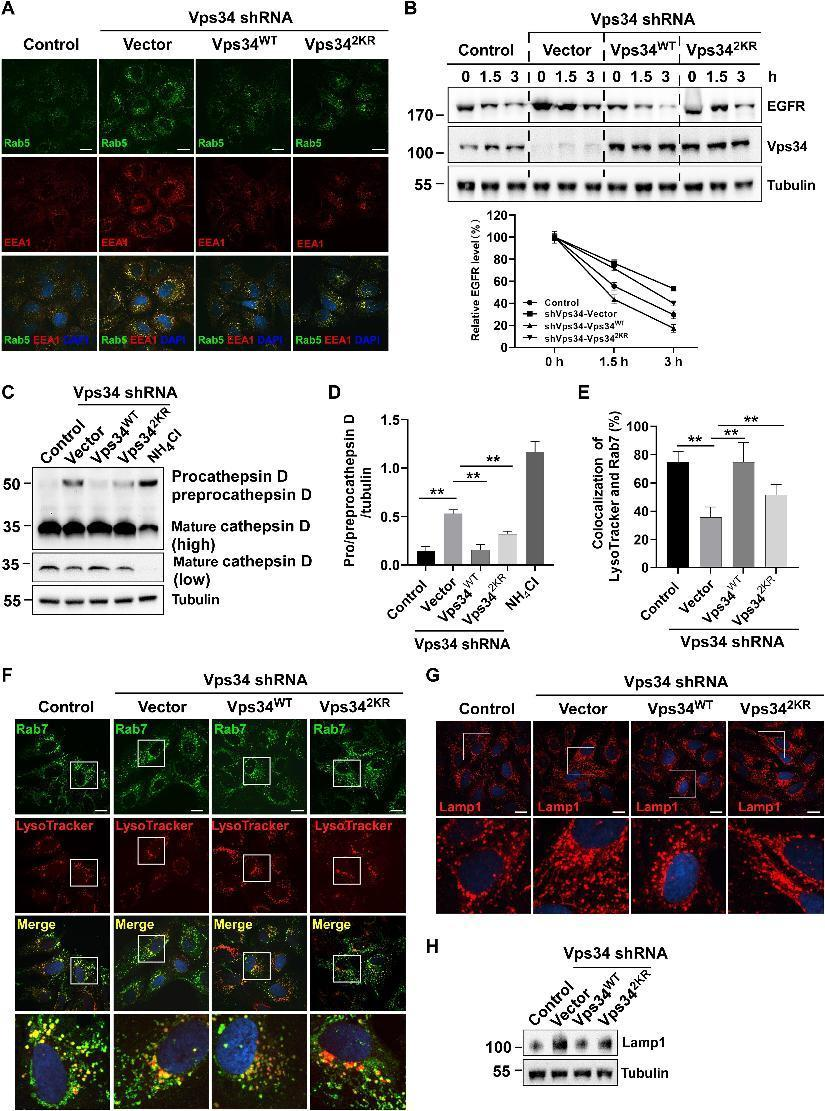
图6:Vps34乳酰化促进内体-溶酶体降解。
7.Vps34的乳酰化在骨骼肌内环境稳定中起重要作用
为了评估Vps34乳酰化的生理功能,构建了强迫游泳小鼠模型,发现游泳3h不影响总AMPKα、总ULK1、葡萄糖转运蛋白4的表达水平游泳诱导AMPKα(Thr172)、总mTOR、ULK1(Ser555)、LDHA(Ser196)的上调和mTOR(Ser2448)和ULK1(Ser757)的下调(图7A和B)。此外,运动诱导小鼠肌肉组织中的乳酸产生,并且该增加被草胺盐处理部分地减少(图7B)。骨骼肌生物化学的评估显示,游泳诱导细胞自噬,如通过p62降解的测量和LC3斑点测定所确定的,并且值得注意的是,这些表型的获得被草胺盐处理部分抑制(图7C-F)。游泳促进Vps34乳糖化并增加PtdIns(3)P水平,并且这些表型被草胺盐处理部分抑制(图7C,D和G)。这些结果表明,通过ULK1-LDHA途径的乳酸盐介导的Vps34乳酸化是骨骼肌稳态所需的。为了获得Vps34乳酰化参与体内自噬和骨骼肌稳态的进一步证据,通过肌内注射用对照病毒rAAV-Vps34 WT或rAAV-Vps342KR(1 × 1011 CFU)处理小鼠。病毒注射4周后,每组半数小鼠进行3小时的急性游泳运动。结果显示,在肌肉中施用Vps 34 WT比对照增强了静息状态和游泳状态下的自噬水平,但施用Vps 342KR通过p62降解和LC3转换比Vps 34WT部分地损害肌肉自噬水平(图7H)。透射电子显微镜分析显示,Vps34WT过表达促进小鼠肌细胞在静息状态和游泳状态下自噬空泡的形成,但Vps342KR降低其作用(图7I和J)。这些结果表明Vps34乳酰化是骨骼肌细胞自噬和细胞稳态所必需的。
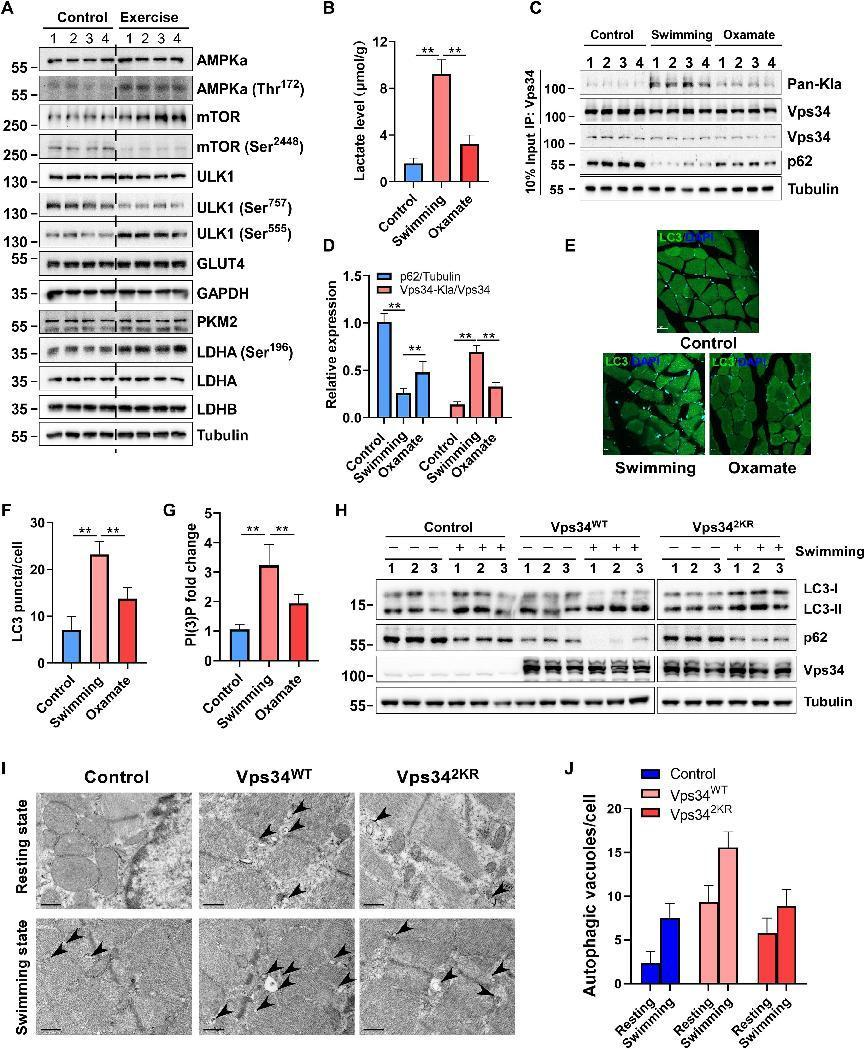
图7:Vps34的乳酰化在骨骼肌内环境稳定中起重要作用
8.Vps34乳糖化与癌症进展相关
为了获得Vps34乳酰化的病理学功能,分析了人肺癌和胃癌中乳酸和Vps34乳酰化水平之间的关系。与邻近组织相比,人肺癌和胃癌中乳酸水平升高(图8A和E)。此外,与邻近组织相比,人肺癌和胃癌组织中Vps34乳酰化程度增加,并且这种乳酰化增加伴随着LDHA表达的上调和自噬通量的增加(图8B,C,F和G)。此外,与邻近组织相比,人肺癌和胃癌组织中的Vps34激酶活性增强,如用体外PtdIns(3)P水平测定所测定的(图8D和H)。结果表明,乳酸通过Vps34乳酰化调节肿瘤组织中的自噬活性,并与癌症进展相关。

图8:Vps34乳糖化与癌症进展相关
结论
总之,这些结果表明,丝氨酸/苏氨酸激酶ULK1通过磷酸化LDHA在S196,以增强LDHA活性和促进乳酸的产生来调节糖酵解途径。乳酸介导的Vps34乳酰化促进其脂质激酶活性以促进细胞自噬和内溶酶体降解。Vps34乳糖化通过调节细胞自噬在运动和肿瘤进展期间的骨骼肌稳态中起关键作用。本研究描述了自噬调控机制,整合了两个高度保守的生命过程(糖酵解和自噬)。
实验方法
收集临床样本,游泳训练动物模型,免疫印迹分析,免疫荧光实验,质谱分析,透射电镜,自噬分析,LDHA酶测定实验,体外乳酰化测定,Vps34激酶测定,PtdIns(3)P ELISA测定,酶标仪检测活细胞中的乳酸盐和NADH/NAD+,流式细胞仪测定活细胞中的乳酸和NADH/NAD+,PAS染色,PCR
参考文献
Jia, M., Yue, X., Sun, W., Zhou, Q., Chang, C., Gong, W., Feng, J., Li, X., Zhan, R., Mo, K., Zhang, L., Qian, Y., Sun, Y., Wang, A., Zou, Y., Chen, W., Li, Y., Huang, L., Yang, Y., Zhao, Y., … Cheng, X. (2023). ULK1-mediated metabolic reprogramming regulates Vps34 lipid kinase activity by its lactylation. Science advances, 9(22), eadg4993. https://doi.org/10.1126/sciadv.adg4993