






自噬基因ATG16L1的肿瘤内在表达抑制了结直肠癌的抗肿瘤免疫

近年来,免疫检查点抑制剂(ICI)如抗PD-1、PD-L1和CTLA-4的单克隆抗体已被证明在许多癌症类型中具有临床疗效。然而迄今为止,只有一小部分结直肠癌(CRC)患者对ICI有应答。因此,揭示MSS-CRC中ICI耐药的决定因素对于通过新型治疗组合解锁抗肿瘤免疫至关重要。炎症性肠病的全基因组关联研究揭示了与肠道炎症增加相关的关键通路。其中,调节caspase介导的降解的核心自噬基因ATG16L1的一个错义变异体被确定为克罗恩病的高度显著风险等位基因,以及CRC14和胃癌的推测预后因素。ATG16L1是自噬分解代谢过程所需的e3连接酶样自噬体延伸复合体的关键成分。自噬在肠上皮细胞适应性中发挥重要作用。此外,自噬通过多种机制调节固有和适应性免疫,包括抗原呈递、效应T细胞功能和调节程序性细胞死亡。最近使用肿瘤细胞系模型的研究表明,核心自噬通路的成员是免疫抑制的肿瘤内源性或外源性决定因素,但它们的转化相关性仍不清楚。该研究发表在《Nature Communications》,IF:16.6。
技术路线

主要研究结果
1. ATG16L1表达升高预测携带致癌性KRAS突变的CRC患者免疫治疗反应差
为了评估ATG16L1表达是否与非MSI CRC对ICI的应答相关,研究者评估了IMblaze370的肿瘤基因表达数据。研究者比较了抗PD-L1单药治疗与抗PD-L1与MAP激酶通路抑制剂考比替尼联合治疗。瑞戈非尼单药治疗被用作标准治疗对照组。
对非高MSI肿瘤中的ATG16L1转录水平所做的分析表明,在KRAS突变型肿瘤中,在阿替利珠单抗和阿替利珠单抗+考比替尼联合治疗组中,ATG16L1表达水平升高均与较差的总生存期相关,但在KRAS野生型肿瘤中未见相关(图1a)。相反,在瑞戈非尼组中,ATG16L1转录水平与有差异的结局无关(图1a)。ATG16L1表达与肿瘤上皮标签呈正相关,与免疫和间质标签呈负相关,提示ATG16L1表达的主要来源是肿瘤细胞(图1b)。IMblaze370肿瘤样本的组织病理学评估显示,ATG16L1蛋白水平主要在肿瘤上皮内升高,少量位于肿瘤间质(图1c)。人CRC肿瘤的单细胞基因表达分析证实,与CRC肿瘤微环境的其他细胞区室相比,肿瘤上皮中的ATG16L1转录水平升高(图1d,e)。
接下来,研究者研究了KRAS状态是否与ATG16L1低表达和高表达肿瘤的转录谱差异相关。使用CIBERSORT40进行的免疫细胞去卷积分析显示,ATG16L1低表达的肿瘤显示了KRAS突变环境中T细胞和NK细胞浸润增加的明显更强证据(图1f)。因此,在KRAS突变患者中,接受免疫治疗的ATG16L1低表达患者的生存改善与T细胞和NK细胞浸润增加相关。综上,在非MSI CRC中,ATG16L1低水平肿瘤患者可能会产生更炎症的微环境,从而在阿替利珠单抗检查点抑制时促进产生高效的抗肿瘤免疫应答。
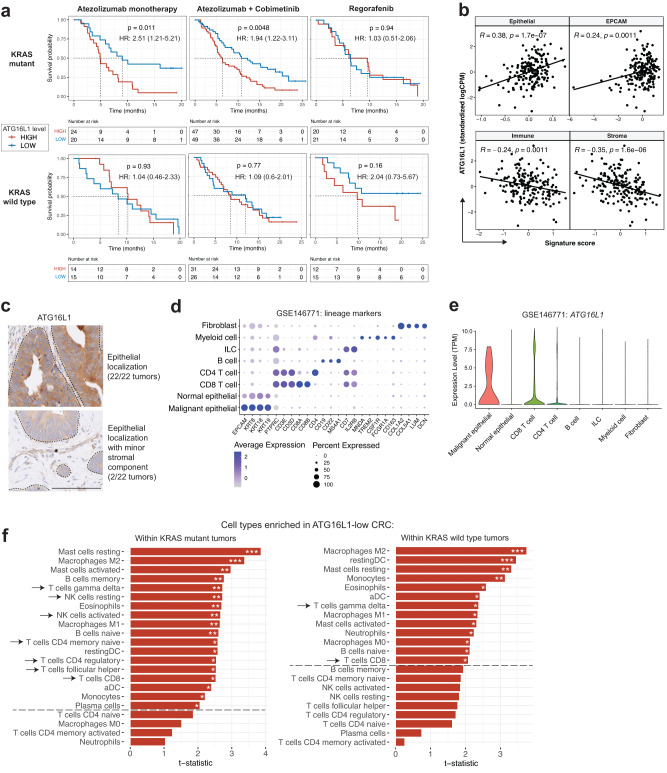
图1 在携带致癌性KRAS突变的非高微卫星不稳定型结直肠癌患者中,ATG16L1表达升高与免疫治疗的不良结局相关
2. 肿瘤内源性ATG16L1促进体内细胞免疫抵抗
在晚期CRC中,ATG16L1表达水平升高与不良结局相关,这一观察结果促使研究者从功能上评估了其在CRC进展中的作用。首先,通过对小鼠结肠类器官进行CRISPR-Cas9编辑,研究者开发了携带在MSS-CRC中观察到的关键驱动突变的肿瘤模型。研究者依次引入了肿瘤抑制因子Apc、Trp53、Smad4的功能缺失和Kras的致癌功能获得,以生成转化的CRC类器官(AKPS,图2a)。ATG16L1通过脂化LC3/ATG8家族的泛素样蛋白16来驱动自噬体延伸。在ATG16L1敲除的CRC类器官克隆中,研究者证实了脂质化LC3b的完全缺失(图2b)。一类称为sequestosome样受体(SLRs)的蛋白质的累积是自噬流缺陷和ATG16L1缺失的标志。与lc3-ⅱ缺失一致,与对照(WT)CRC类器官相比,ATG16L1敲除(KO)中SLRs SQSTM1/p62和CALCOCO1水平升高(图2b)。
结直肠癌转移主要发生在肝和肺。研究者分别使用改良的水动力尾静脉注射方案和传统的静脉尾静脉注射将CRC类器官直接输送到肝脏和肺。将CRC类器官原位注射到结肠上皮中用于在原发疾病龛中建立肿瘤。通过生物发光(BLI)成像观察稳定表达的荧光素酶报告基因在肝和肺模型中的肿瘤生长情况。在完全免疫正常的宿主中,ATG16L1缺失显著降低了肝脏肿瘤负荷(图2c,d,i)。接种后6周,给予WT CRC类器官的小鼠的肝脏显示出广泛的疾病负担,而给予ATG16L1 KO CRC类器官的少数小鼠仅出现偶尔的肿瘤灶(图2i)。总之,研究者的数据表明ATG16L1促进免疫正常小鼠的CRC生长,而不依赖于组织生态位。由于IMblaze370研究了转移后CRC的患者结局,因此研究者的后续实验侧重于CRC类器官的肝脏定植。
ATG16L1缺失不影响体外AKPS类器官的基础生长,这提示肿瘤内在的ATG16L1缺失可能引发体内控制疾病的肿瘤外源性机制。研究者将WT和ATG16L1 KO CRC类器官植入免疫受损宿主的肝脏(图2e-h)。NOD/SCID-gamma IL2Rgnull(NSG)小鼠是研究人类肿瘤细胞在体内生长的常用小鼠。与细胞免疫在控制肿瘤生长中的作用一致,野生型CRC类器官在NSG宿主中比免疫正常的BL6小鼠生长更快。值得注意的是,ATG16L1 KO CRC类器官在NSG宿主中快速生长(图2e,h),并且在免疫正常的对照BL6宿主中达到与WT CRC类器官相同的程度(图2d)。虽然ATG16L1 KO组的肿瘤负荷与WT组相比仍然显著降低,但给予ATG16L1 KO CRC类器官的所有NSG小鼠均表现出较大的肿瘤结节(图2j)。
为了进一步完善这些观察结果,研究者分别去除宿主细胞毒性T淋巴细胞或自然杀伤(NK)细胞,然后植入CRC类器官。有趣的是,耗竭NK细胞显著恢复了ATG16L1 KO CRC类器官的生长,而耗竭CD8+T细胞无显著影响(图2f,h)。因此,NK细胞似乎是清除ATG16L1缺陷的MSS-CRC的关键因素。
接下来,研究者使用Ifng KO小鼠研究IFNγ是否参与肿瘤控制,IFNγ是驱动细胞毒性T细胞和NK细胞效应功能的关键细胞因子。与野生型对照宿主相比(图2d),如BLI定量所示,宿主IFNγ缺失显著增加了ATG16L1 KO CRC类器官的肝脏定植(图2g,h),提示宿主IFNγ在体内抑制ATG16L1缺陷型CRC类器官的生长。总体而言,研究者观察到ATG16L1缺陷的CRC类器官在免疫受损宿主的肝脏中生长改善(图2h)。研究者的结果表明,不依赖于组织生态位,肿瘤固有的ATG16L1极大地限制了CRC的免疫介导控制。而当肿瘤在免疫缺陷宿主中生长时,其影响相对较小。
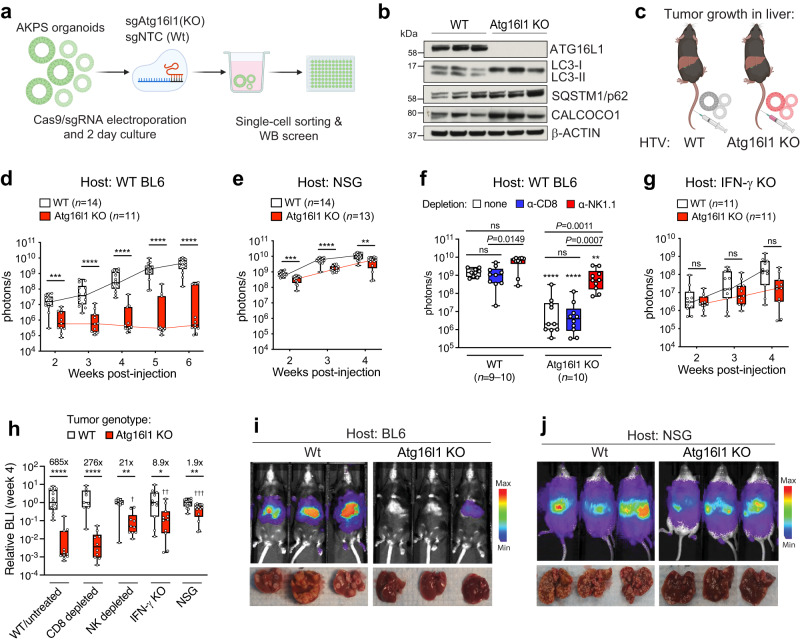
图2 ATG16L1通过促进细胞免疫抵抗来驱动肝内CRC生长
3. ATG16L1的缺失改变了CRC类器官的组成和表型
为了更好地理解ATG16L1如何影响肿瘤及其微环境中细胞的表型编程,研究者对植入NSG小鼠肝脏中的肿瘤进行了转录组学分析,因为ATG16L1 KO CRC类器官的最佳生长需要免疫缺陷宿主。收集肿瘤并将活细胞分选为CRC类器官和白细胞组分。通过单细胞RNA测序分析每组的分选部分。对类器官细胞和免疫细胞的数据分别进行预处理和标准化。将类器官细胞和免疫细胞池分别过滤,仅保留上皮细胞和髓样细胞。
研究者首先聚焦于肿瘤固有状态,其中无监督图聚类揭示了上皮CRC细胞的8个簇:增殖(Prolif.),分泌/感觉(SS),成熟肠细胞(ME),神经内分泌(NE),肠祖细胞,干扰素反应(IFN),StemHI和Goblet/Paneth(GP)细胞簇(图3a,b)。
细胞类型比例的比较表明,ATG16L1缺失显著改变了体内CRC类器官细胞的组成(图3c-e)。在ATG16L1 KO组中,IFN反应性CRC细胞的比例显著增加,指向依赖自噬的肿瘤固有免疫抑制过程(图3e)。ATG16L1 KO组的NE和SS反应簇也呈比例增加(图3e)。相比之下,在ATG16L1 KO组中,GP细胞的比例显著降低,突显了ATG16L1在维持杯状细胞和潘氏细胞身份方面的重要作用(图3e)。最后,ATG16L1缺失导致StemHI和增殖细胞簇成比例减少(图3e)。
ATG16L1缺失也改变了每个簇内CRC类器官细胞的转录状态。基因集富集分析显示,除了显著增加的IFN反应簇外,在ATG16L1 KO组中,StemHI和GP细胞簇也表现出更高水平的IFN反应基因,这与炎症性肿瘤微环境的产生、肿瘤对IFN的内在敏感性增加或两者的某种组合一致(图3f)。此外,在ATG16L1缺失后,除IFN应答簇外,所有簇均显示上皮-间充质转化(EMT)相关基因水平升高(图3f)。考虑到与EMT样特征密切相关的NE簇的比例增加,这一结果凸显了EMT是ATG16L1 KO条件中最显著的变化之一。以上结果表明,即使在免疫功能低下的NSG宿主中,TME也可能通过增强IFN反应对ATG16L1缺陷肿瘤施加选择性压力。
研究者接下来的问题是,在体内植入的ATG16L1缺陷肿瘤中观察到的转录变化中,哪些是真正的肿瘤固有的。为了严格关注ATG16L1的肿瘤内在效应,研究者对体外培养的WT和ATG16L1 KO CRC类器官进行了批量RNA-seq分析。对单细胞来源的细胞簇的前100个标记物进行GSEA分析,发现ATG16L1缺失导致Prolif的表达显著降低。和GP簇标志物,以及SS簇标志物表达增加(图3g,h),表明这些是肿瘤细胞中ATG16L1缺失导致的直接变化。相比之下,其他簇的变化显然是由TME介导的(图3e-g)。总之,这些数据提示,ATG16L1缺失导致CRC类器官中的杯状细胞、帕内特细胞、干细胞和增殖池减少,可能抑制肿瘤生长。同时,增强的IFN反应性可能增加抗肿瘤免疫压力。
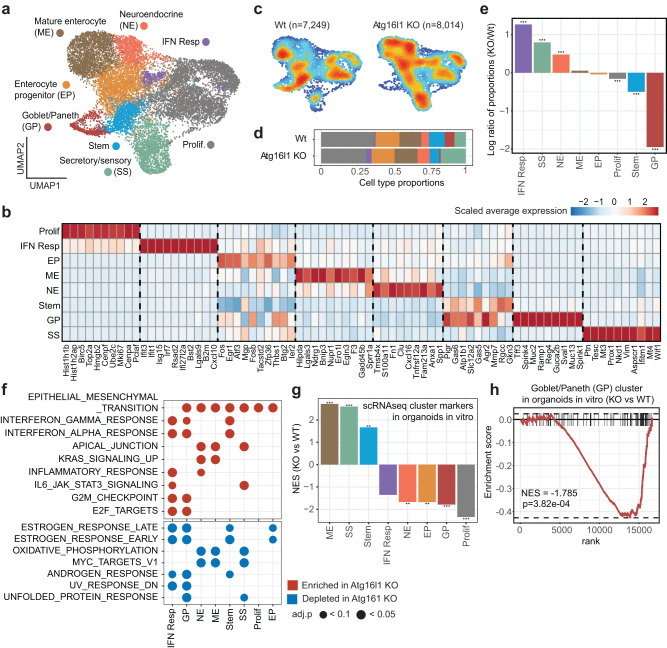
图3 ATG16L1的肿瘤内在缺失改变了肝脏中CRC细胞的表型和组成
4. 结直肠癌类器官中ATG16L1的缺失重塑了肿瘤髓系隔室
接下来,研究者探讨了肿瘤内在ATG16L1缺失如何影响造血肿瘤微环境。由于ATG16L1 KO CRC类器官在免疫正常的BL6宿主中未形成肿瘤,因此研究者的分析仅限于NSG宿主中生长的肿瘤的髓系室。在本研究中,研究者对NSG小鼠肝脏内种植的WT和ATG16L1 KO肿瘤分选的髓系细胞进行了scRNA-seq分析,这些髓系细胞分为10个簇(图4a,b)。巨噬细胞亚群包括TREM2+、VSIG4+、MARCO+和ifn反应性巨噬细胞/单核细胞的混合簇。树突状细胞聚集成cDC1、cDC2、CCR7+DC和浆细胞样DC。另外两种不同的细胞群是单核细胞和中性粒细胞。在CRC类器官中ATG16L1的缺失导致了髓系室的实质性重塑。ATG16L1 KO组单核细胞以及TREM2+和VSIG4+巨噬细胞亚群显著减少(图4c-e)。ATG16L1缺失后,中性粒细胞募集增强,这与即使在NSG宿主中炎症活动增加一致(图4e)。正如无偏倚GSEA分析中IFN反应特征的增加所表明的那样,大多数巨噬细胞和DC亚群显示了复极化进入炎症状态的证据(图4f)。IFN反应的混合巨噬细胞/单核细胞簇的百分比显著增加(图4e),并且在ATG16L1 KO条件下表现出更强的IFN反应(图4f)。这些结果表明,肿瘤固有的ATG16L1缺失不仅重编程CRC细胞,而且导致髓系室向抗肿瘤状态的促炎重塑。
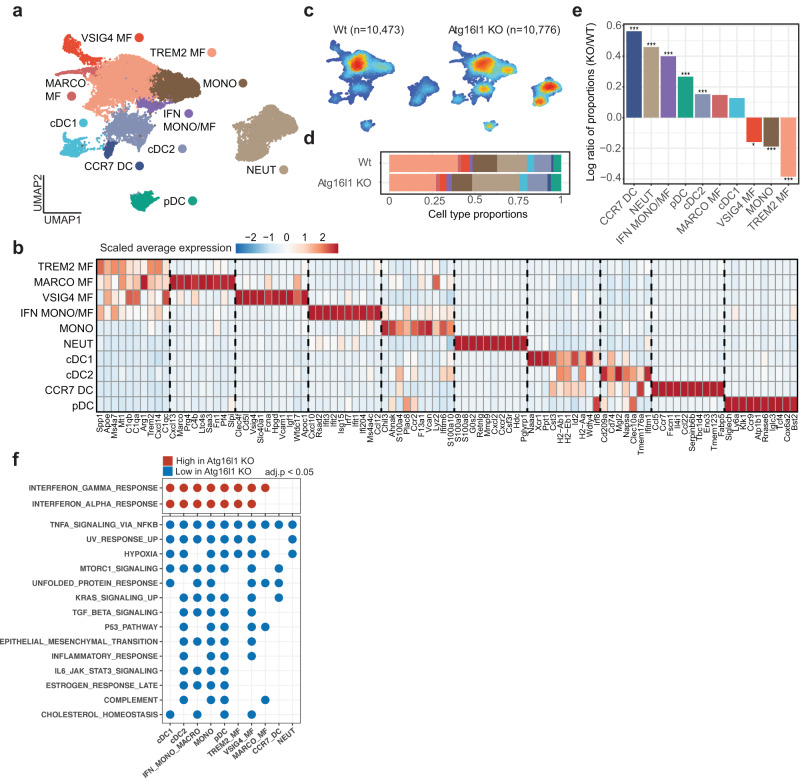
图4 肿瘤内ATG16L1缺失可重编程肿瘤微环境中的髓系细胞组成和表型
5. ATG16L1保护CRC类器官免受TNF+ifnγ介导的程序性细胞死亡
细胞毒性淋巴细胞和IFNγ对ATG16L1 KO肿瘤的清除增加,以及NSG小鼠中ifn应答基因标签的持续增强,促使研究者直接确定肿瘤内源性ATG16L1在调节IFNγ信号传导中的作用。首先,在体外IFNγ刺激下对野生型和ATG16L1 KO型结直肠癌类器官进行基因表达谱分析。在IFNγ处理的CRC类器官中,ATG16L1的缺失显著增强了IFN通路基因的表达(图5a,b)。之前的研究表明,在高度转化的细胞系或原代肠上皮细胞中,自噬抑制使其对TNF或IFNγ诱导的细胞毒性敏感,与之相反,IFNγ或TNF单独在体外无法诱导CRC类器官死亡(图5c,d)。然而,TNF和IFNγ联合使用可诱导CRC类器官死亡。重要的是,ATG16L1的缺失显著加速了TNF和IFNγ共同处理诱导的细胞死亡(图5c,d)。
接下来,研究者研究了参与细胞因子介导的细胞死亡的信号通路。有趣的是,重要的程序性坏死介质RIPK3和混合谱系激酶结构域样蛋白仅在ATG16L1 KO类器官中被磷酸化(图5e)。在ATG16L1 KO CRC类器官中,Ripk3缺失部分挽救了TNF+ifnγ诱导的死亡,在ATG16L1和Ripk3双重缺失的细胞中添加z-VAD可完全阻断细胞死亡(图5f)。抑制ATG16L1 KO类器官中的半胱氨酸天冬氨酸蛋白酶可进一步加速细胞死亡,这可能是通过RIPK3介导的程序性坏死实现的,这与半胱氨酸天冬氨酸蛋白酶在程序性坏死中的调节作用一致。总之,这些结果表明ATG16L1的缺失使CRC类器官对TNF+ifnγ诱导的程序性坏死和凋亡敏感。
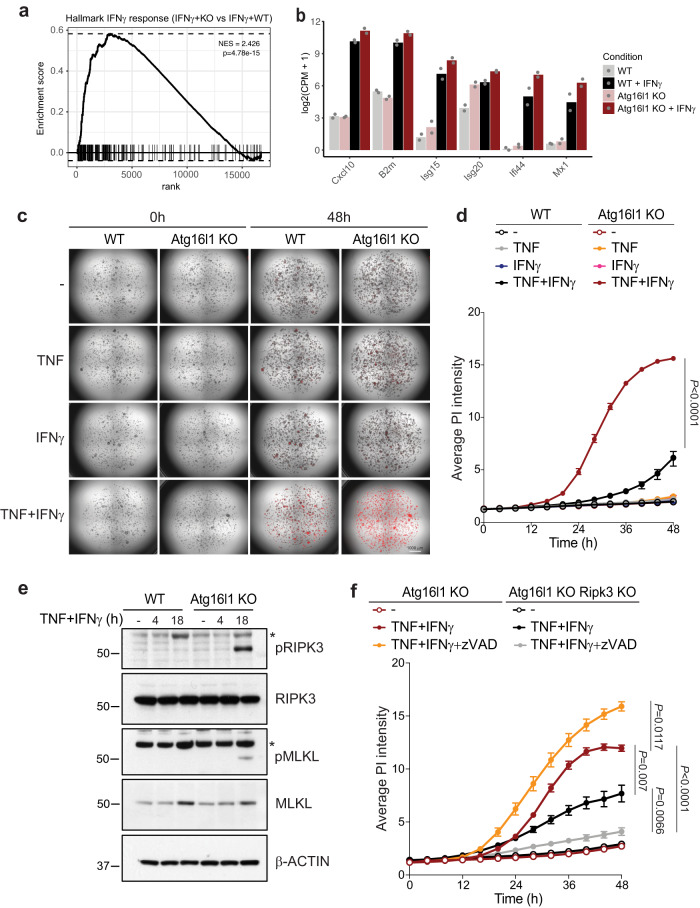
图5 ATG16L1缺失使CRC类器官对TNF+ifnγ诱导的细胞死亡敏感
结论
该研究表明,CRC中ATG16L1的高表达预示着较差的免疫治疗应答,这在机制上可以通过ATG16L1介导的IFN信号传导抑制和随后的细胞免疫抑制来解释。这为在晚期MSS-CRC中通过抑制自噬来克服免疫疗法耐药提供了治疗理论依据,而MSS-CRC是一种可选择的治疗方案有限、临床结局不佳且未满足的需求高的疾病。
实验方法
生物发光成像,蛋白质印迹,流式细胞术,细胞死亡测定,免疫组织化学染色,全外显子组测序,TCGA分析,生存分析,单细胞RNA测序
参考文献
Taraborrelli L, Şenbabaoğlu Y, Wang L, Lim J, Blake K, Kljavin N, et al. Tumor-intrinsic expression of the autophagy gene Atg16l1 suppresses anti-tumor immunity in colorectal cancer. Nat Commun. 2023 Sep 23;14(1):5945. doi: 10.1038/s41467-023-41618-7.