






缺乏长寿基因会加重骨关节炎?!
高度保守的依赖NAD+的sirtuin脱乙酰酶和单-ADP核糖基转移酶家族(sirtuins 1-7)是关键的表观遗传调节因子,在各种模式生物中控制与年龄相关的细胞信号通路并延长寿命。自从发现小鼠体内Sirt6的整体缺失导致孕激素表型、代谢障碍和出生4周内死亡以来,阐明核局部sirtuin 6(SIRT6)在衰老和疾病中的确切作用的努力已经成为当务之急。相反,在雄性和雌性小鼠中,Sirt6的转基因过表达控制衰老过程中的代谢信号事件,从而延长寿命。一些证据表明,Sirt6调节一系列与年龄相关的生物过程,包括DNA修复、细胞代谢、氧化应激、炎症、自噬和衰老。
年龄和关节损伤是骨关节炎(OA)的关键风险因素,骨关节炎是最常见的关节疾病,也是老年人残疾的主要原因。在软骨细胞中,年龄相关或损伤相关的改变有利于分解代谢而不利于合成代谢的信号事件,这促进了细胞外基质(ECM)成分的丢失,并被认为是OA发展和进展中驱动软骨降解的原因。最近的证据表明,SIRT6可能是这些过程的关键调节因子。例如,体外细胞培养研究表明,SIRT6过表达降低了人软骨细胞的复制衰老、基质金属蛋白酶-13水平和核因子-kappaB(nf-ĸB)调控的基因表达,而SIRT6缺失则增加了软骨细胞中DNA损伤和端粒功能障碍诱导的病灶的标志物,并显著抑制了软骨肉瘤SW1353细胞系中COL2A1和ACAN基因的表达。在小鼠体内的数据表明,Sirt6单倍体功能不全会增加软骨蛋白多糖丢失和膝下脂肪垫细胞因子水平,导致高脂饮食的中年小鼠的国际骨性关节炎研究学会(OARSI)得分更高。此外,在胶原诱导和K/BxN血清转移的类风湿性关节炎模型中,小鼠骨髓特异性Sirt6缺陷已被证明可增加关节炎症和对前分解代谢FOXO1信号事件的敏感性,从而增强关节炎症。相反,在接受内侧半月板去稳定(DMM)手术的年轻小鼠中,关节内给予腺相关病毒或慢病毒SIRT6,以增加关节间隙内的SIRT6水平,提供对软骨损伤的保护。
我们以前在原代人软骨细胞中的研究表明,SIRT6的激活通过增加抗氧化蛋白水平、降低促氧化剂TXNIP水平和快速解毒核产生的H2O2来促进对氧化应激的抗性。此外,激活SIRT6显著减少氧化应激诱导的分解代谢NF-ĸB信号事件,这些事件与软骨细胞死亡和骨关节炎有关。重要的是,我们的报告还显示,在人类关节软骨细胞中,软骨细胞SIRT6活性随着年龄的增长而显著下降。然而,关节内SIRT6缺乏的影响,以及这如何导致体内软骨损伤和骨关节炎,在很大程度上仍未被研究。由于先前研究Sirt6体内作用的研究使用了少量实验小鼠(n = 4–6),本研究的目的是全面确定Sirt6缺乏对OA的影响。由于损伤和年龄是OA的两个主要危险因素,我们研究了Sirt6缺乏对年轻小鼠的影响,以内侧半月板手术作为创伤后OA的模型,并评估了中年(12个月龄)和老年(18个月龄)小鼠自发、自然发生的OA的严重程度。SIRT6调节软骨细胞功能以保护软骨免受OA侵袭的具体机制在这些小鼠中以及在体外使用原代人软骨细胞进行了检测。本文于2023年8月发表于《TRANSLATIONAL SCIENCE》IF=27.4期刊上。
技术路线
主要结果
1.软骨特异性Sirt6缺乏增加DMM诱导的小鼠骨关节炎的严重程度
因为小鼠Sirt6的整体缺失导致4周龄内死亡,我们建立了可诱导的软骨特异性Sirt6缺陷小鼠(Sirt6fl/fl;Aggrecan-CreERT2,(Sirt6 cKO)),并将它们与Sirt6完整的同窝对照组(Sirt6fl/fl)进行比较。Sirt6完整和Sirt6缺陷小鼠在16周龄时接受DMM手术或假手术,并在术后6周和10周通过组织学、详细的组织形态计量学和显微CT分析OA的严重程度。在组织学上,接受DMM手术的Sirt6完整和Sirt6缺陷小鼠都出现了OA的症状,其特征是在研究的两个时间点与假对照组相比,关节软骨结构(ACS)、Toloniumchloride、骨赘和滑膜增生评分显著增加(图1A-D)。当比较DMM组(Sirt6完整与Sirt6 cKO)时,与术后6周和10周Sirt6完整小鼠相比,Sirt6缺陷小鼠的ACS、Toloniumchloride和骨赘评分明显更高(更差)(图1A-D)。
与我们的组织学数据一致的是,对胫骨平台内侧和外侧的详细组织形态学分析表明,接受假手术的小鼠几乎没有显示出OA的迹象,而DMM手术在所研究的两个时间点都产生了OA表型,这由显著的软骨损失以及在某些情况下钙化软骨的损失和软骨下骨板(SCBP)面积和厚度的增加所证明(表1和2)。手术后6周,与接受DMM手术的Sirt6完整小鼠相比,接受DMM手术的Sirt6缺陷小鼠的关节软骨面积和厚度显著减少(表1)。在第10周时,这种OA表型恶化,并且与经历DMM的Sirt6完整小鼠相比,软骨特异性Sirt6缺乏的小鼠也表现出钙化软骨厚度的显著减少和SCBP面积和厚度的增加(表2)。
微型CT分析胫骨软骨下骨体积分数(Bv/Tv)、骨小梁厚度(Tb.Th)、骨小梁间距(Tb.Sp)和软骨下骨板厚度(SCBP)。在所有小鼠的胫骨内侧平台上进行Th。在手术后6周,接受DMM手术的Sirt6基因缺陷小鼠的BV/Tv和Tb.Th显著增加,Tb.Sp值显著降低,这表明与接受假手术的SIRT6基因缺陷小鼠相比,这组小鼠的骨硬化程度增强(图1E和G)。在这一时间点,来自两个手术组的Sirt6完整小鼠之间没有观察到任何变化。术后10周,与假手术组相比,两个DMM组的BV/Tv和Tb.Th均增加,Tb.Sp显著降低,表明DMM在这个时间点诱导了骨硬化(图1F和H)。在分析DMM组之间的差异时,Sirt6缺陷小鼠与Sirt6完整小鼠相比,BV/Tv和Tb.Th显著增加,Tb.Sp减少,这表明在没有Sirt6的情况下骨硬化增强(图1F和H)。接受DMM手术的Sirt6缺陷小鼠的骨赘面积和骨赘体积也显著大于Sirt6完整小鼠(图1I,J),这与我们的组织学骨赘评分一致。总之,这些数据表明,小鼠软骨中Sirt6缺乏增加了DMM诱导的骨性关节炎的严重程度。
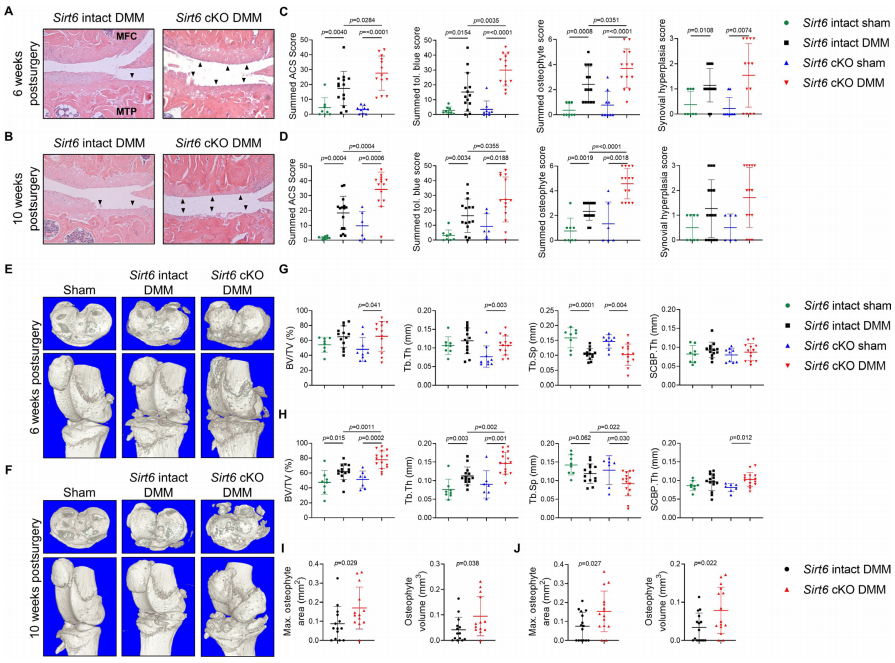
图1 Sirt6缺乏对DMM术后骨性关节炎严重程度的影响
表1 DMM术后6周Sirt6完整和Sirt6缺陷(Sirt6 CKO)小鼠接受DMM或Sham手术的组织形态计量学分析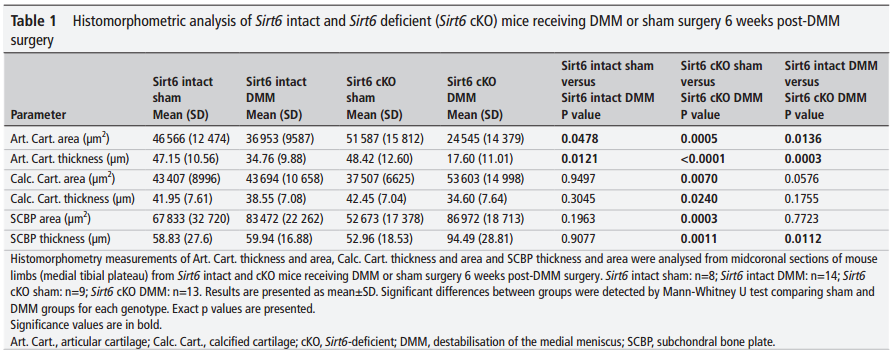
表2 DMM术后10周Sirt6完整和Sirt6缺陷(Sirt6 CKO)小鼠接受DMM或Sham手术的组织形态计量学分析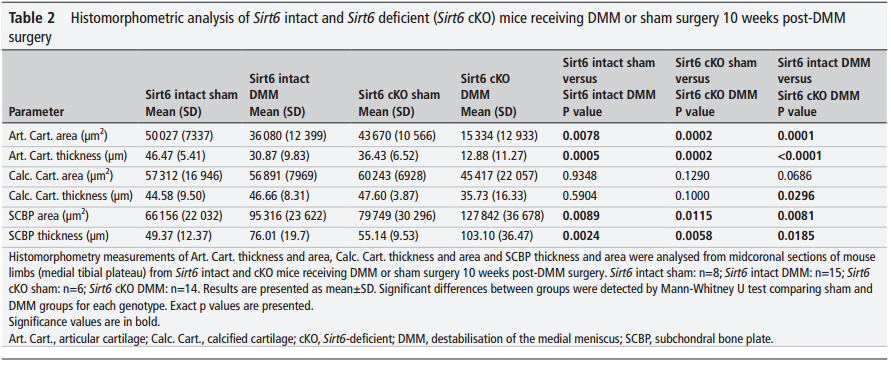
2.软骨特异性Sirt6缺乏加速小鼠自发的年龄相关性骨性关节炎严重程度
为了评估Sirt6缺乏对自发、自然发生的骨性关节炎的影响,对12个月和18个月龄的Sirt6完整和Sirt6缺乏的小鼠进行了骨性关节炎的严重程度分析,根据DMM研究。在12个月大时,与Sirt6完整对照组相比,Sirt6缺陷小鼠的ACS、Toloniumchloride和骨赘总分显著增加(图2A,B)。一致的是,详细的组织形态计量学分析显示,与Sirt6完整的对照组相比,Sirt6缺陷小鼠的内侧和外侧关节软骨面积和钙化软骨面积都显著减少(表3和4)。在小鼠18个月时,两种基因型都表现出严重的骨性关节炎,在胫骨平台内侧有明显的软骨丢失,但两种基因型之间没有显著差异(图2,表3)。在外侧,18个月龄的Sirt6缺陷小鼠的关节软骨面积和厚度与Sirt6正常对照相比显著减少(表4)。在这项老龄研究中,年龄或基因对滑膜增生没有影响(图2B)。同样,当我们在两个时间点分析来自两个基因型的四肢时,我们没有通过Micro-CT检测到软骨下骨参数(内侧)的任何显著差异(图2E,F)。滑膜增生和软骨下骨改变不受年龄的影响,这一发现与我们之前在类似时间点评估这些参数的小鼠研究一致。对18月龄Sirt6完整小鼠和Sirt6基因缺陷小鼠的关节组织切片进行免疫组织化学检测,以检测作为衰老标志的p16ink4a。与正常对照组相比,Sirt6基因缺陷小鼠滑膜中p16ink4a阳性细胞显著增加(p=0.0031)。综上所述,这些数据表明,软骨特异性Sirt6缺乏显著加速了小鼠自发的、与年龄相关的骨性关节炎。
有趣的是,当分析6个月大的DMM组(假手术后10周)的对照小鼠肢体,并将它们与12个月和18个月大的完整Sirt6对照进行比较时,我们观察到BV/Tv和Tb.Th值增加,而Tb.Sp值在老年小鼠肢体中降低。这一发现表明,小鼠的衰老本身就会增加软骨下骨硬化症,据我们所知,这是一个原创性的发现。
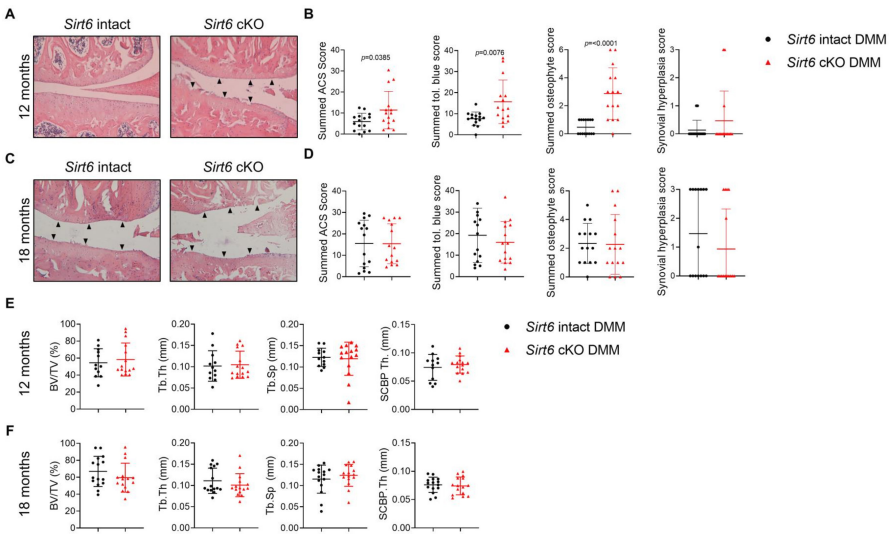
图2 SIRT6缺乏对衰老过程中骨性关节炎严重程度的影响
表3 12月龄和18月龄Sirt6完整和Sirt6缺陷(Sirt6 CKO)小鼠(胫骨内侧平台)的组织形态计量学分析

表4 Sirt6完整和Sirt6缺陷(Sirt6 CKO)小鼠12月龄和18月龄(胫骨外侧平台)的组织形态计量学分析

3.Sirt6缺陷与人软骨细胞ECM和生长因子基因下调有关
为了确定Sirt6介导的转录调控的作用,对缺失Sirt6的原代人软骨细胞进行了RNA测序,并与干扰的小干扰RNA(SiRNA)对照细胞(72小时)进行了比较。初步研究证实SIRT6 siRNA缺失,并证明与对照组相比,SIRT6 siRNA的核导入导致SIRT6蛋白水平显著降低(p=<0.0001)。在我们的RNA测序数据集中,主成分分析表明,按治疗方法分组。SIRT6缺失的样本与对照组的比较显示,236个基因差异表达,其中160个基因下调,76个基因上调(图3A)。分析表明,软骨细胞SIRT6的缺失预计会增加关节炎症、软骨损伤和OA疾病的进程。基因本体论浓缩分析表明,与对照组相比,细胞外基质、蛋白质类细胞外基质(细胞成分)和生长因子活性(分子功能)术语是缺失SIRT6的人软骨细胞中最受抑制的三个过程(图3B)。
对差异表达的ECM基因的查询显示,在关节软骨中发现的主要胶原COL2A1在SIRT6缺失的细胞中与其他ECM基因如COMP、ECM2、CILP2和COL8A1一起显著减少。qRT-PCR法显示,与对照组相比,SIRT6缺陷软骨细胞中COL2A1(p=0.0008)和comp(p=0.0001)基因的表达显著降低(图3C)。在生长因子抑制的背景下,促合成胰岛素样生长因子-1(IGF1;p=0.0070,IGFBP2;p=0.0011)在SIRT6缺失的软骨细胞中表达显著下调(IGF1;p=0.0070,IGFBP2;p=0.0011)。上游调控分析表明,IGF-1的下调预计会抑制我们数据集中发现的各种促合成软骨基因,包括COL2A1和IGFBP2(图3D)。在核糖核酸测序数据集中其他显著下调的基因包括Sirt6(p=0.0011),Wnt抑制物SFRP1(p=0.0048)和SFRP4(p=0.0002),以及著名的SIRT6调节的代谢和寿命调节因子PCK1(p=0.005)(图3C)。相反,缺乏SIRT6导致促进分解代谢的IL1RL1和HHIP基因显著增加,当增强时,这两个基因与OA的进展和发展有关。这些效应也通过定量逆转录聚合酶链式反应(IL1RL1;p=0.0121,HHIP;p=0.0008)得到验证(图3A和C)。
综上所述,这些结果表明,SIRT6的缺失显著降低了IGF-1基因和包括COL2A1在内的一系列ECM基质基因的表达,并促进了与OA相关的促分解代谢基因的基因表达。由于IGF-1信号在维持软骨细胞外基质和软骨细胞存活中起着关键作用,这些结果促使我们进一步探索SIRT6/IGF-1轴在软骨细胞中的作用。
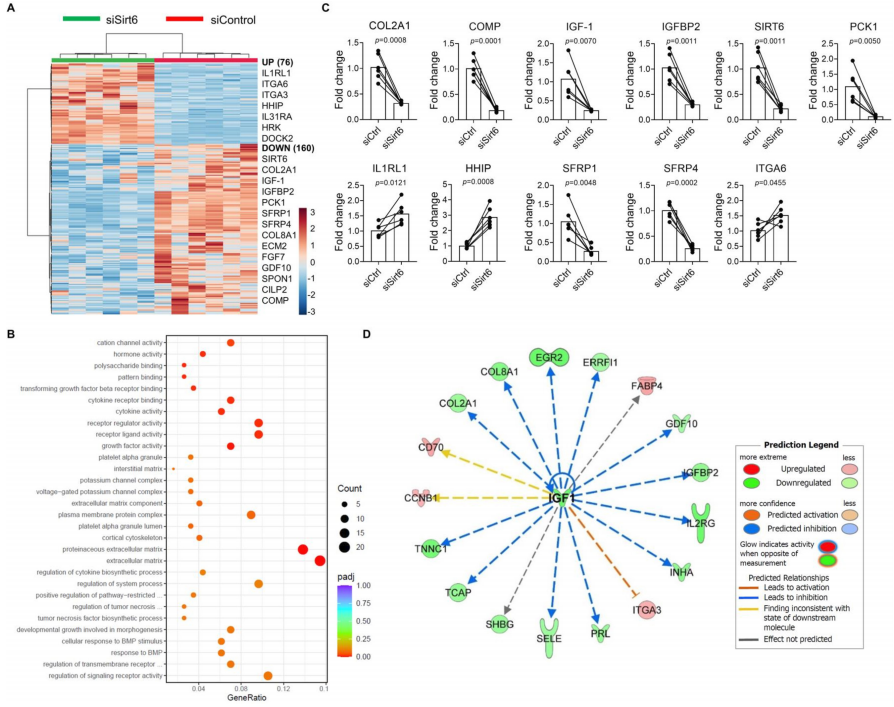
图3 Sirt6缺失的人软骨细胞的RNA测序分析
4.SIRT6对人软骨细胞中IGF-1信号的调控
为了评估Sirt6缺陷对IGF-1信号转导的影响,本研究对Sirt6完整和Sirt6缺陷小鼠的股骨头软骨进行了解剖和体外培养。用4-羟基他莫昔芬处理外泌体96小时,以诱导Cre介导的Sirt6缺失,然后提取软骨细胞并进行免疫印迹处理,以检测IGFBP2和磷酸化的Akt,作为IGF-1信号通路激活的标志。与Sirt6完整的股骨头外植体相比,缺失Sirt6的外植体显示基础IGFBP2(p=0.0112)和磷酸化Aktser473(p=0.0229)蛋白水平显著降低(图4A)。对来自我们的数字MM研究中的Sirt6完整和Sirt6缺失的假对照组小鼠的关节组织切片进行的免疫组化研究表明,与Sirt6完整的对照组相比,Sirt6缺陷的小鼠软骨中胰岛素样生长因子-1的水平显著降低(p=0.0002),这与我们在Sirt6基因敲除的人软骨细胞中的RNA测序数据一致(图4B)。
由于Sirt6缺乏降低了小鼠软骨中的IGF-1/Akt轴,我们接下来的目标是测试SIRT6激活对软骨细胞中IGF-1信号转导的影响。用编码SIRT6的腺病毒载体(24小时)转导原代人软骨细胞以增加SIRT6的活性,或如前所述的空载体(空)对照。与空载体对照相比,SIRT6过表达显著增加了Akt在Ser473(p=0.0178)和Thr308(p=0.0003)处的基础磷酸化,并增加了Akt活性的标志--富含Pro的Akt底物的磷酸化(p=0.0249)(图4C)。接下来,我们用SIRT6的小分子激活剂MDL800处理原代人类软骨细胞,此前已有研究表明,它可以将SIRT6的活性提高22倍。我们分离了软骨细胞组蛋白,并对SIRT6底物的乙酰化形式H3K9(H3K9ac)进行了免疫印迹,H3K9ac是SIRT6活性的反向标记。用MDL-800(12.5微米,24小时)处理软骨细胞后,H3K9的基础乙酰化形式显著减少(p=0.0001),表明与对照组相比,SIRT6活性增强。在总细胞裂解物中,MDL800诱导的SIRT6激活导致基础磷酸化Aktser473和磷酸化PRAS40显著增加,并在处理3小时时达到峰值(分别为p=0.0170,p=0.0120)(图4D)。在研究的时间过程中,SIRT6蛋白水平没有随着MDL-800的处理而改变,这与其他研究结果一致。总之,这些功能获得和功能丧失的研究表明,SIRT6是小鼠和人类软骨细胞中促合成代谢的IGF-1信号通路的关键调节因子,并减少与骨关节炎相关的分解代谢信号事件。
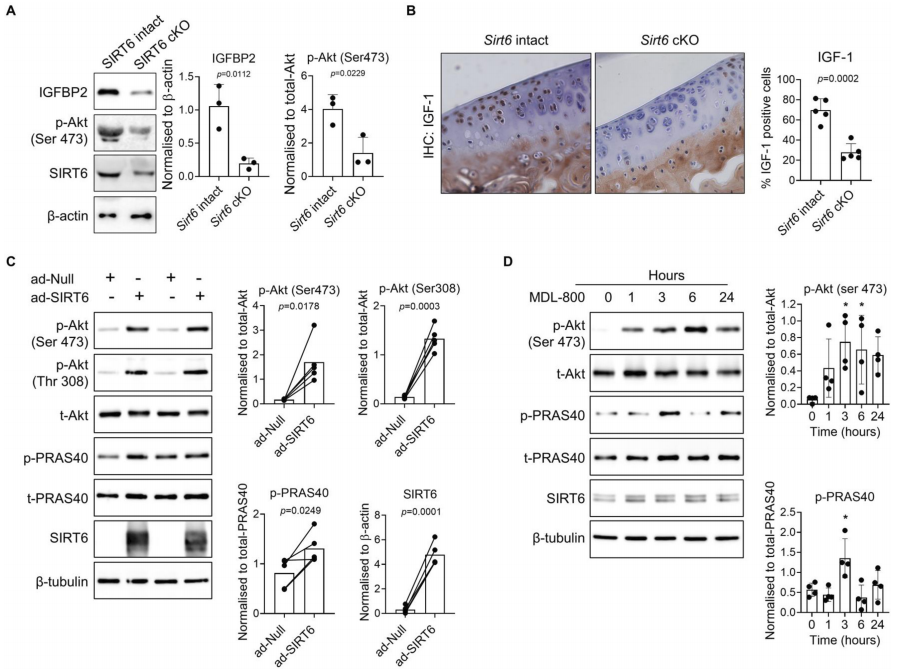
图4 SIRT6调节软骨细胞中的胰岛素样生长因子-1(IGF-1)信号
实验方法
H&E染色、免疫印迹、RT-qPCR
参考文献
Collins, J. A. et al. Cartilage-specificSirt6deficiency represses IGF-1 and enhances osteoarthritis severity in mice. Annals of the Rheumatic Diseases, 2023 doi:10.1136/ard-2023-224385 (2023).