






CD36+癌相关成纤维细胞通过分泌巨噬细胞迁移抑制因子为肝癌提供免疫抑制微环境
肝细胞癌(HCC)是全球癌症相关死亡的第四大原因,慢性乙型肝炎(HBV)病毒感染是主要危险因素。超过80%的HCC病例的特征是由成纤维细胞的激活、增殖和积累引起的广泛肝纤维化。HCC肿瘤微环境(TME)的一个显著特征是大量的癌相关成纤维细胞(CAFs),它可以分泌多种细胞因子、趋化因子和生长因子,直接或间接地支持癌细胞。在以往的研究中,CAF在人胰腺导管腺癌(PDAC)、头颈部鳞状细胞癌、乳腺癌和肺肿瘤中广泛发现了不同的CAF亚型,并具有不同的功能。因此,CAFs的多面性表明它们包括不同的亚群,并且对基质异质性的更好理解可以解释CAFs如何促进肿瘤生态系统的动态复杂性和功能可塑性。作者的研究结果提供了一个全面的转录组学概述,并揭示了CD36+ CAFs, CD33+ MDSCs和HCC细胞之间新的交互,为HCC提供了一个潜在的微环境靶向治疗方式。本文于2023年3月发表于《Cell Discovery》,IF=33.5。
技术路线

主要研究结果
1、单细胞分析揭示了人类HCC肿瘤的复杂性
为了全面表征人类HCC中存在的细胞群,作者采用scRNA-seq方法对原发肿瘤中的大量细胞进行分析。来自未经治疗的HBV相关HCC患者的7个肿瘤和其中4个患者的邻近肝组织被酶酶消化以产生单细胞悬液(图1A;110658细胞)。严格过滤后,保留90572个细胞作进一步分析。在基因表达归一化之后,作者分别使用主成分分析和均匀流形近似和投影(UMAP)进行了降维和聚类(图1B)。利用已知的标记基因,细胞可以划分为9种不同的细胞类型(图1B):上皮细胞和肿瘤细胞(5059个细胞,5.6%,用EPCAM、ALDH1A1和ALB标记);B细胞(1332个,1.5%,标记有MS4A1和CD79A);T细胞24895个,占27.5%,CD3D和CD3E标记;自然杀伤细胞(NK) 13868个,占15.3%,标记FGFBP2和FCG3RA;髓源性抑制细胞(MDSCs)(11087个细胞,12.2%,用ITGAM和CD33标记);单核细胞或巨噬细胞(9978个,占11.0%,标记为CD68、CD163和CD14);树突状细胞(7784个,8.6%,用ITGAX标记);成纤维细胞(2495个,2.8%,标记有ACTA2和COL1A2);内皮细胞(14074个,占15.5%,标记有PECAM1和vWF;图1C)。点图所示的差异表达基因(DEGs)和标记基因证实了准确性(图1C)。
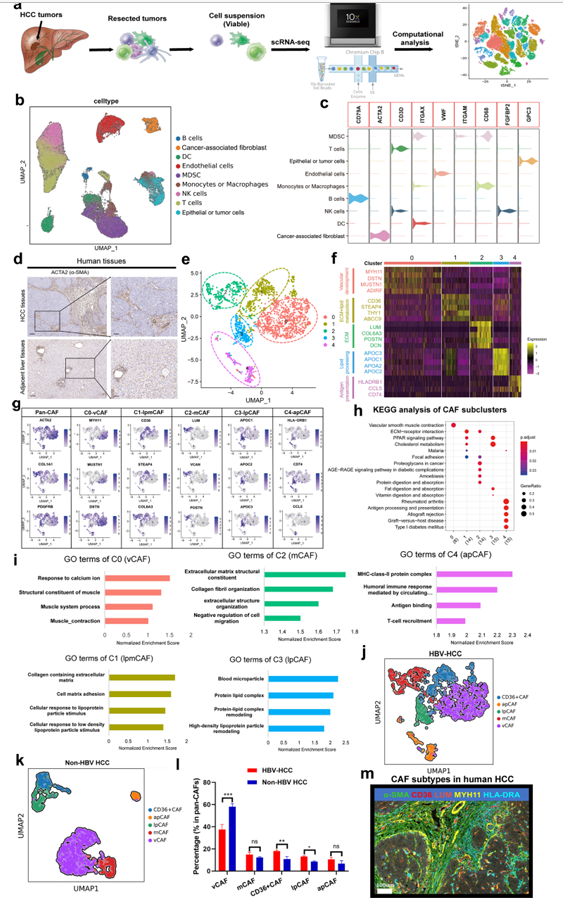
图1. 人HBV相关HCC中不同的成纤维细胞亚群
2、人类HCC中5种CAF亚型的鉴定和分子特征
α-SMA免疫组化(IHC)染色显示,超过80%的HCC病例表现为由成纤维细胞激活和积累引起的广泛肝纤维化(图1D)。作者在来自7个HBV相关HCC组织的scRNA-seq分析中对1835个CAFs进行了分类,并将这些细胞聚集成5个亚群(图1E)。所有5个亚簇都表达高水平的典型成纤维细胞标记,如ACTA2 (α-SMA)、COL1A2和COL1A1;然而,每个亚簇显示出不同的转录组特征(图1F、G)。亚群0 CAF占CAF群体的大部分(40%),其特征是微血管特征基因,如MYH11、MUSTN1和MCAM(图1F、G)。因此,作者将其命名为血管CAFs (vCAFs, vCAFs-c0- myh11)。vCAFs的GO和KEGG分析表明,血管平滑肌收缩和对钙离子的反应显著富集,与它们的微血管特征一致(图1H、I)。亚簇2 CAFs表达低水平的α-SMA和高水平的细胞外基质(ECM)特征,包括胶原分子(COL5A1和COL6A3)、骨膜蛋白(POSTN)、LUM、DCN和FAP(图1F、G)。有趣的是,GO分析表明ECM和胶原原纤维组织相关显著富集。因此,作者相应地将它们命名为基质CAFs (mCAFs, mCAFs-c2-LUM,图1H、I)。与mCAFs-c2-LUM类似,亚群1 CAFs也表达高水平的COL6A3和COL1A1以及高水平的脂质代谢相关基因(CD36和STEAP4,图1F、G)。此外,该亚簇显著富集于ECM、胆固醇代谢、脂肪酸代谢标志和活性氧(ROS)途径相关(图1H、I),表明该亚簇可能参与ECM和胆固醇代谢。因此,该亚簇中的CAFs被命名为脂质加工(lp)-mCAFs (lpmCAFs, lpmCAFs-c1- CD36;图1F、G)。亚群3 CAFs表达高水平的脂质加工标志物,包括APOA1和APOC1(图1G)。有趣的是,GO和KEGG分析表明,这些CAFs在蛋白质-脂质复合物重塑和脂肪酸代谢标志中显著富集;因此,作者将它们命名为脂质加工CAFs (lpCAFs, lpCAFs-c3- APOA1;图1H、I)。与之前报道一致,作者发现亚群4 CAF表达主要的组织相容性复合体II (MHCII)基因,如CD74和HLA-DRA,以及趋化因子相关基因,如CCL5(图1F、G)。此外,在该亚簇中富集的GO项和KEGG通路与MHCII类蛋白复合体和抗原加工和呈递有关(图1H、I);因此,作者称之为抗原呈递CAFs (apCAFs, apCAFs-c4- HLA-DRA)。最后,为了探讨不同病因导致的CAF亚型的异质性,作者对7例非HBV相关HCC肿瘤进行了scRNA测序,发现与HBV-HCC相比,非HBV HCC(包括酒精型、脂肪肝或药物性HCC)中CD36+ CAF和lpCAFs的比例显著降低(图1J、I),这可能是HBV相关HCC中HBV蛋白诱导的脂质代谢和氧化应激引起的。最后,作者使用多重免疫荧光(mIF)染色证实了人类HCC样本中主要CAF亚群的存在(图1M)。
3、小鼠HCC肿瘤中CAF亚型的单细胞分析概括了人类HCC中发现的亚型
作者在人类HCC标本中的结果证实了5个CAF亚簇的存在。为了更深入研究CAF,作者将研究扩展到小鼠HCC模型。作者之前的研究显示,在作者的HBV相关HCC患者队列中,CTNNB1(58%)和TP53(19%)是两个突变最多的基因。因此,作者建立CTNNB1N90;Trp53KO HCC小鼠模型,模拟HCC遗传背景(图2A)。在此模型的基础上,将来自HCC小鼠的7个肿瘤分离成单个细胞,经过严格过滤,共获得63977个活细胞(图2B)。对该数据集进行聚类分析得到10个簇,中位数为6054个转录本,每个簇有2262个基因(图2B)。与人类HCC相似,小鼠HCC肿瘤含有大量髓系细胞(约30%),主要由巨噬细胞和MDSCs组成,还有一小部分DC。成纤维细胞在HCC肿瘤中的比例与人类HCC样本相似(占所有细胞的2.7%)。为了进一步辨别成纤维细胞的异质性,作者在作者的scRNA-seq分析中对来自7个小鼠HCC肿瘤的1746个CAFs进行了分类,并将这些CAFs聚类为7个亚群(图2C)。所有7个亚簇都表达高水平的典型成纤维细胞标记物,如Col1a2和Col1a1(图2E);然而,每个亚簇显示出不同的转录组特征(图2D、E)。有趣的是,作者发现在人类HCC肿瘤中鉴定的所有5种CAF亚型在小鼠HCC组织中也存在(图2C)。此外,5个小鼠CAF亚群具有与人类CAF相似的群体和基因特征,包括vCAFs、mCAFs、CD36+CAFs、lpCAFs和apCAFs。最后,作者使用特异性标记物,包括Myh11、Lum、Acta2、Cd36和H2-Aa进行mIF染色,以证实主要CAF簇也存在于小鼠HCC组织中(图2G)。
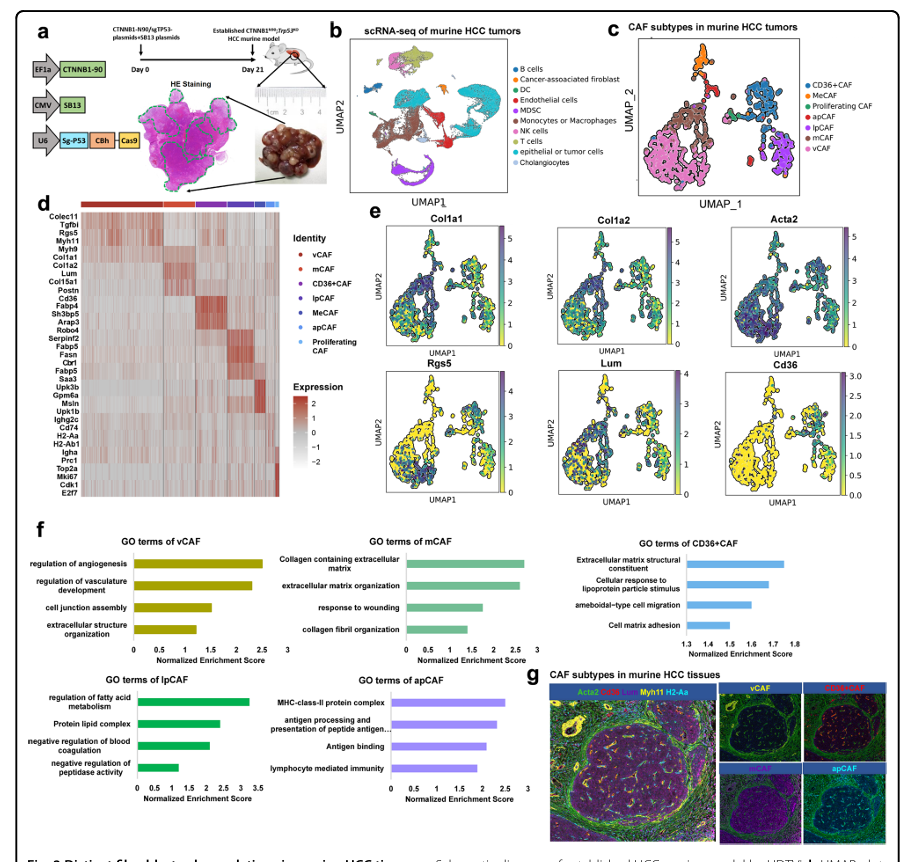
图2. 小鼠HCC组织中不同的成纤维细胞亚群
4、CAF亚型特异性转录因子和基因调控网络
接下来,作者试图鉴定TFs及其靶向基因调控网络,以更好地了解CAF亚型是如何在遗传上建立和维持的。为此,作者应用软件SCENIC来识别在一种CAF亚型中高度活跃的TFs及其靶标。作者观察到vCAFs在MEF2C和FOS中富集(图3A、B),之前认为MEF2C和FOS可以调节新生血管生成。此外,MEF2C的靶基因,如MYLK、ACTA2和MYH11,在vCAFs中上调(图3D)。TWIST1和CREB3L1是mCAFs中表达量最高的TFs(图3A、B), TWIST1是CAF转分化的关键因子。此外,TWIST1的靶基因(COL1A1, MMP2)在mCAFs中上调最多(图3D)。lpmCAFs (CD36+ CAFs)显示CEBPD的高表达(图3A、C), CEBPD是一种已知的控制与脂质代谢和EMT相关的转录程序的关键调节因子。脂蛋白脂肪酶(LPL)靶基因在lpmCAFs中上调(图3D)。除CEBPD外,lpCAFs还高表达CEBPA和MAFB(图3A、C),这两种已知的TFs参与促进LDL代谢转录程序。最后,apCAFs高表达IKZF1和RUNX3(图3A、C),它们与巨噬细胞极化和T细胞募集有关。为了检测在这些小鼠CAF亚群中具有活性的主调控因子,进行了SCENIC分析,结果显示Mef2c和Foxf1是小鼠vCAFs中的活性TFs基因,与人类vCAFs类似(图3E、F)。同样,Twist1、Cebpd、Cebpa和Ikzf1也分别是小鼠mCAFs、CD36+ CAFs、lpCAFs和apCAFs中的活性TFs基因(图3E)。总的来说,这些结果确定了在已确定的CAF亚型中驱动或维持基因表达程序的关键TFs,进一步深入了解HCC肿瘤中CAF异质性的基因调控网络。
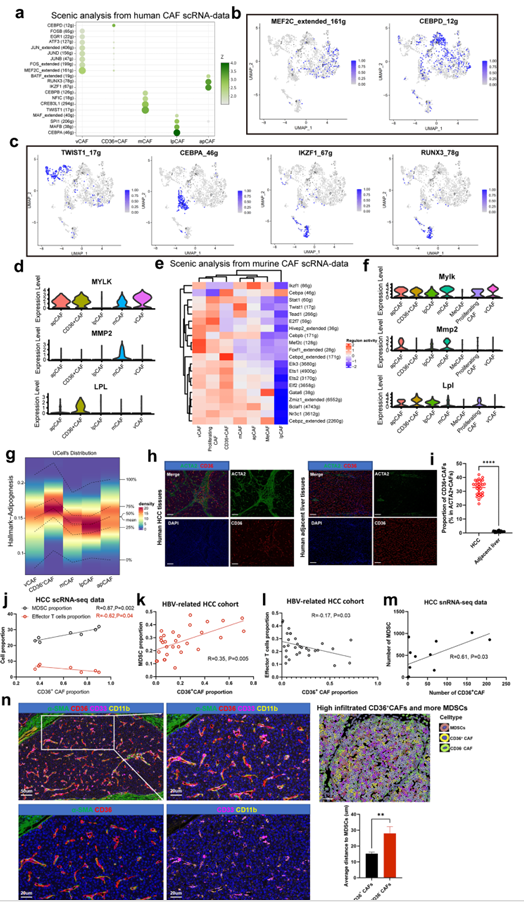
图3. CAF亚型特异性TF以及不同CAF与MDSCs之间的相互作用
5、CD36+ CAF与MDSCs呈正相关,并且在HCC肿瘤中与MDSCs关系密切
在人类或小鼠HCC肿瘤中确定了CAF亚型后,作者下一步探索了CAF与TME中其他细胞之间的特定串扰机制。值得注意的是,在所有已知的配体-受体对中,MIF信号通路由MIF配体及其CD74/CXCR4受体主导。有趣的是,通常和高度表达CD36的lpmCAFs和lpCAFs是作用于MDSCs的MIF配体的最主要来源。进一步的免疫组化实验表明CD36的表达主要位于HCC间质区。因此,作者使用特异性表面标记物在lpmCAFs (c1-mCAF-CD36)和lpCAFs这两个亚群中鉴定了CD36,发现它们都在脂肪形成途径中富集(图3G),占HCC中CAF群体的35%,表明它们在HCC进展中起关键作用。进一步的mIF实验表明,与小鼠和人体组织的邻近肝组织相比,CD36+ CAFs在肿瘤组织中特异性浸润(图3H、I)。首先,作者研究了CD36+ CAFs与TME内的效应T细胞MDSCs之间是否存在相关性。作者的scRNA-seq数据和TCGA数据显示CD36+ CAFs和MDSCs之间呈正相关(图3J),但观察到效应T细胞(GZMB+ CD8+)呈负相关(图3J),这一点在30例原发性HBV相关病例的石蜡包埋肿瘤切片中的IF检测中得到了验证(图3K-I)。此外,对12例HCC肿瘤进行单细胞核测序(snRNA-seq),发现CD36+ CAFs的数量与MDSCs呈正相关(图3M)。接下来,作者验证了CD36+ CAFs和MDSCs之间的相互作用。mIF染色显示CD36+ CAFs比CD36 - CAFs更接近MDSCs(图3N)。总的来说,这些结果表明CD36+CAFs可能在HCC中具有有效的免疫抑制作用。
6、流式细胞术分离肝癌细胞CAF亚型
为了分析CAF亚群,作者检查了单细胞数据,并鉴定了在每个CAF亚群中独特表达的表面蛋白。使用CD36、CD146、FAP和MHCII抗体,将CAFs分离成四组细胞:(1)CD36阳性、(2)MHCII阳性、(3)FAP阳性和(4)CD146阳性细胞,推测分别对应于CD36+ CAFs、apCAFs、mCAFs和vCAFs(图4A)。流式细胞术分析显示,在CD31和EpCAM双阴性人群中,小鼠和人类HCC肿瘤中CAF亚型的比例大致相似(图4B)。为了验证该策略是否适用于CAF分选,作者通过FACS分离细胞,并进行qPCR分析。所有四个CAF亚群均高表达泛成纤维细胞标记物Col1a1、Col2a1和Acta2(图4C)。CD36阳性CAF的独特之处在于它们高表达Cd36、Fabp4和Sh3bp5,以及lpCAF标记物Fabp5、Apoc1和Fasn(图4C)。MHCII阳性的CAFs在Cd74和H2-Ab1的高表达方面是十分显著的(图4C)。FAP阳性细胞显示mCAF标记物Lum、Col5a1、Col6a3、Timp2、Mmp2和Twist1的相对高表达(图4C)。Mcam阳性的cas显示vCAF标记Myh11、Myh9、Mustn1和Rgs5的高表达(图4C)。最后,FAP阳性细胞显示mCAF趋化因子(如Lum、Mmp2和Mmp1)的相对表达显著升高,表明该群体中mCAFs富集(图4C)。这些结果表明,Lrat谱系间质细胞是小鼠HCC发展过程中CD36+ CAFs的主要来源。
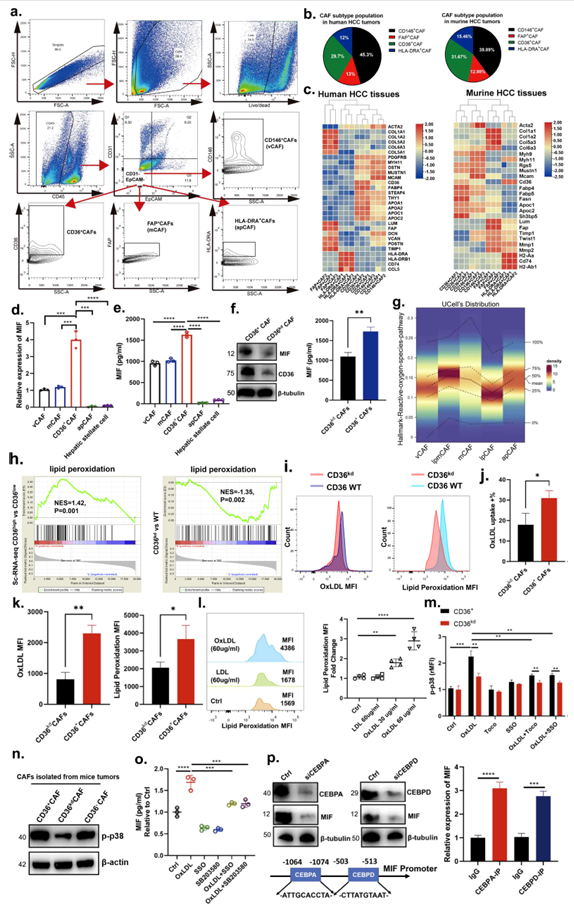
图4. CD36介导OxLDL摄取,通过脂质过氧化/p38/ cebp轴促进MIF在CD36+ CAFs中的表达
7、CD36介导OxLDL摄取,通过脂质过氧化/p38/ CEBP轴促进MIF在CD36+ CAFs中的表达
为了检测不同CAF产生的MIF水平,我们通过FACS分离不同CAF亚型。进一步的qPCR和ELISA实验表明,来自CD36+ CAFs的MIF表达明显高于来自vCAFs、mCAFs、apCAFs和hsc的MIF表达(图4D、E)。基于这些结果,我们随后将重点放在CD36+ CAFs的这一亚群上进行进一步的研究。我们还从HCC组织中分离和扩增了CD36+ CAFs。Western blotting和ELISA分析显示,当CD36在CD36+ CAFs中沉默时,CD36+ CAFs表达和分泌的MIF减少(CD36kd CAFs;图4F),表明MIF的表达可能受到CD36的调控。重要的是,这些结果表明CD36+ CAFs可能通过旁分泌信号促进MDSCs中某些促肿瘤功能的获得。CD36是一种清道夫受体,在脂质代谢中起作用,并被报道参与炎症反应和代谢紊乱,如糖尿病和肥胖。在免疫系统中,CD36已被报道介导树突状细胞抗原获取和呈递,并支持调节性t细胞功能。然而,人们对其在caf中的作用知之甚少。与我们通过CAF scRNA-seq分析确定的信号通路相似(图4),我们发现CD36+ cas比CD36kd cas具有更高的脂质过氧化相关基因表达(图4H)。使用荧光偶联OxLDL和流式细胞术,我们发现CD36+ CAFs比CD36kd CAFs具有更高的OxLDL摄取率(图4I、J)。基于BODIPY 581/591 C11脂质过氧化测定,CD36+ CAFs与CD36kd CAFs相比,脂质过氧化也显着增加(图4K)。此外,我们想确定OxLDL是否会增加CD36+ CAFs中的脂质过氧化。首先,我们测量了OxLDL对CD36+ CAFs中脂质过氧化的影响,结果显示,OxLDL而非LDL以剂量依赖的方式增强了CD36+ CAFs中的脂质过氧化(图4I)。氧化应激,包括脂质过氧化,可以激活p38激酶及其下游信号通路。因此,我们通过测量CD36+ CAF中p38的磷酸化来检测OxLDL是否可以激活p38。我们发现OxLDL在CD36+ CAF中诱导p38磷酸化,但在CD36kd CAF中诱导的程度要小得多,并且Toco或SSO的添加减少了OxLDL诱导的p38磷酸化(图4M)。此外,体内CD36+ CAFs中p38磷酸化的富集程度高于CD36kd或CD36阴性CAFs(图4N)。这表明OxLDL通过CD36和脂质过氧化促进p38活化。接下来,我们想确定p38是否作用于OxLDL-CD36信号的下游以促进MIF表达;因此,我们在体外用OxLDL处理CD36+ CAFs,无论是否存在p38抑制剂SB203580。结果显示,在OxLDL存在的情况下,p38抑制部分挽救了MIF的分泌(图4O),表明OxLDL部分通过p38激活促进MIF分泌。从我们的SCENIC分析中,我们发现CEBP家族(CEBPA和CEBPD) tf对脂质代谢至关重要,在CD36+ CAFs(包括lpmCAFs和lpCAFs)中特异性富集。先前的研究已经确定p38MAPK是CEBPA和CEBPD激活的关键调节因子。因此,我们首先在CD36+ CAFs中检测了MIF分泌是否依赖于p38诱导的CEBPA和CEBPD激活。当CEBPA和CEBPD分别沉默时,我们观察到MIF在经OxLDL处理的CD36+ CAFs中显著降低(图4p)。然后,我们进行了生物信息学分析,以确定MIF启动子中CEBPA或CEBPD的潜在结合位点(图4P)。接下来,进行染色质免疫沉淀(ChIP)实验,发现CEBPA和CEBPD都可以直接结合到MIF启动子上(图4P)。综上所述,这些结果表明CD36介导OxLDL摄取,通过脂质过氧化/p38/CEBP轴促进MIF表达。
8、CD36+ CAFf衍生的MIF通过IL-6/STAT3激活增强了MDSCs促进免疫抑制性TME和肿瘤干细胞的能力
基于上述结果表明,CAFs的数量与MDSCs的数量相关,作者假设CD36+ CAFf衍生的MIF促进了CD33+ MDSC的扩增。为了确定来自肿瘤细胞或CD36+ CAFs的MIF是否介导CD33+ MDSC扩增的调节,从原发性人HBV相关或小鼠HCC肿瘤中分离人或小鼠CD36+ CAFs。使用CD36+ CAFs (CD36+ CAFs-CM)、CD36 - CAFs、CD36kd CAFs (CD36kd CAFs-CM)、CD36+ CAFs+MIF抑制(ISO-1)或CD74阻断的条件培养基(CM)来处理MDSC前体,并测量CD33+ MDSCs的比例(图5A、B)。发现CD36+ CAFs-CM、MIF增加了CD33+ MDSCs的数量(图5C)。CD36+ CAFc - cm + ISO-1和CD36+ CAFc - cm + CD74阻断在抑制CD33+ MDSC扩增方面表现出良好的活性(图5C),这表明MIF分泌是CD36+ CAFs调控MDSC扩增的关键介质。然后,在免疫功能正常的小鼠原位HCC模型中,作者证明了共注射从小鼠HCC肿瘤中分离的CD36+ CAFs(~30:1)可显著促进小鼠肝脏中HCC肿瘤细胞的肿瘤生长或转移,而特异性敲低CD36、抑制MIF或耗尽Gr-1+ MDSCs可大大减弱这种作用(图5D、E)。共注射CD36+ CAFs可显著增加肿瘤中Treg的浸润,但减少效应CD8+ T细胞的浸润(图5F、G),特异性敲低CD36、抑制MIF或消耗Gr-1+ MDSCs也会大大减弱这种浸润(图5F、G)。这些数据表明,CD36+ CAFs的促瘤作用是通过MDSCs的免疫抑制功能间接介导的。MDSCs可以通过几种不依赖于接触的机制促进免疫抑制,包括诱导型一氧化氮合酶(iNOS)的表达,已知iNOS可抑制实体瘤中CD8+ T和NK细胞的细胞毒性和功能。首先,为了研究CD36+ CAFs是否可以通过iNOS信号传导来教育MDSCs增强其免疫抑制能力,作者从CD36+ CAFs、CD36kd CAFs或CD74阻断细胞的原代CD36+ CAFs、CD33+ MDSCs或经CM预处理的CD33+ MDSCs培养物中收集CM,并评估iNOS水平。CD36+ CAFc -MDSC-CM,而CD36kd + CAFc -MDSC-CM或CD36kd阻断MDSC的CM,显著促进了MDSCs中iNOS的分泌(图5H)。此外,作者的共培养实验显示,与MDSC- wt组相比,CD36+ CAF + MDSC组的CD69+、IFNG+ CD8+ T细胞比例显著下调,但treg和PD1+CD8+ T细胞比例上调,而CD36+ CAF + MDSC + ISO-1或CD74阻断只产生轻微影响(图5I、J)。这表明CD36+ CAFs通过MIF/CD74/ iNOS轴以依赖CD36的方式增强了CD33+ MDSCs的免疫抑制能力。此外,作者使用裸小鼠原位HCC模型证明,共注射CD36+ CAFs可显著促进裸小鼠肝脏中肿瘤细胞的生长或转移,而特异性敲低CD36、抑制MIF或耗尽Gr-1+ MDSCs可极大地减弱肿瘤细胞的生长或转移。这些数据表明,CD36+ CAFs的促肿瘤作用是由MDSCs的非免疫抑制功能间接介导的,可能具有干细胞增强能力。最后,为了探索CD36+ CAFf衍生的MIF影响MDSCs的分子机制,作者进行了RNA测序,并比较了MDSC-MIF组和MDSC组。结果显示,核因子κB (NF-κB)信号通路被列为最重要的途径,并被报道调节MDSCs中细胞因子的分泌,在CD36+ CAFf衍生的MIF预处理的MDSCs中被激活(图5K、I)。为了研究导致CD36+ CAFf相关的MDSC扩张的机械刺激的下游信号,作者首先检测了CD36+ CAFs诱导的MDSCs和来自HCC患者外周血的MDSCs中NF-κB蛋白p65的磷酸化。作者发现CD36+ CAFs诱导的MDSCs中p65的磷酸化增加(图5M)。据报道,p65激活可显著诱导iNOS和IL-6分泌。ELISA结果显示,与对照组相比,CD36+ CAFs或MIF诱导的MDSCs中iNOS和IL-6分泌显著上调(图5N)。
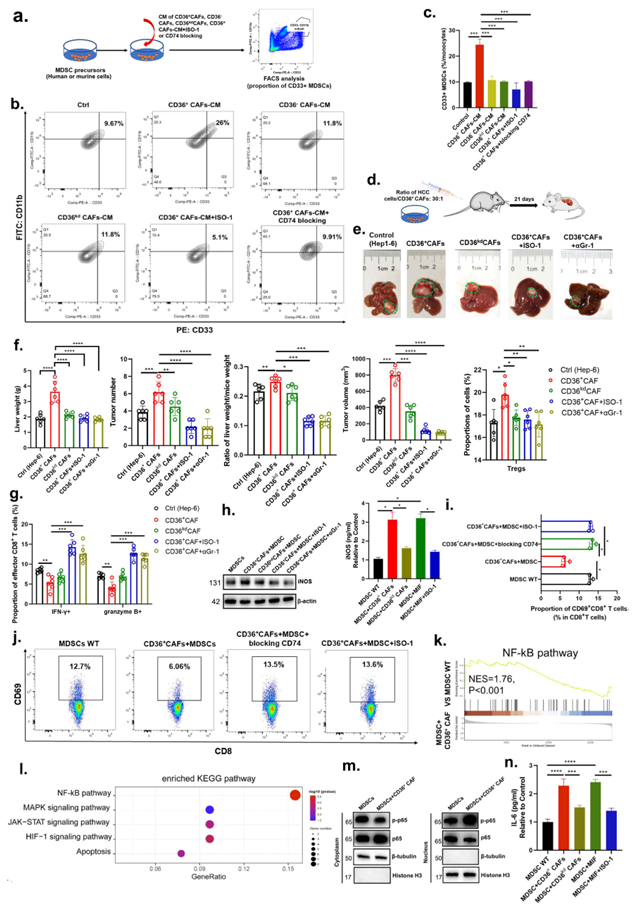
图5. CD36+ CAF衍生的MIF通过IL-6/ STAT3激活增强了MDSCs促进免疫抑制性TME和肿瘤干细胞的能力
9、靶向CD36与免疫治疗在肝癌小鼠模型中的协同作用
根据上述发现,CD36+ CAFf来源的MIF促进了免疫抑制性MDSC的积累,加速了HCC的进展。为了进一步研究CD36+和MIF+ CAFs是否在HCC发生过程中发挥作用,作者建立了CD36 (Acta2Cre; Cd36fl/fl)和MIF (Acta2Cre;Miffl/fl)条件敲除6周龄小鼠,确定在体内敲除CD36或MIF时,HCC肿瘤负荷和MDSCs比例显著降低(图6A-D),表明CD36+ CAFs参与HCC的发生。据报道,单核细胞MDSCs具有免疫抑制作用,与癌症免疫治疗反应差有关。此外,已发现MDSCs诱导的癌症干性可促进多种癌症类型的免疫治疗抵抗。因此,治疗性靶向或减少MDSCs联合免疫检查点阻断剂可以增强t细胞免疫治疗,达到最佳的抗肿瘤效果。为此,作者还假设CD36+ CAFs的数量与HCC患者免疫治疗的疗效有关。因此,作者首先研究了CD36+ CAFs在接受新辅助抗pd -1治疗(托利哌单抗联合lenvatinib作为可切除肝细胞癌的新辅助治疗)的患者切除的HCC组织中的作用;临床试验号:NCT03867370)。结果显示,CD36和αSMA共表达在抗pd -1反应组中低于无反应组,这表明CD36+ CAFs数量少预示着更好的HCC免疫治疗反应(图6E、F)。然后,作者探索了单药(CD36抑制剂或抗pd -1治疗)和联合治疗策略(CD36抑制和抗pd -1治疗)在C57/BJ6 CTNNB1N90;Trp53KO HCC模型和作者建立的抗PD-1耐药HCC中的疗效。有趣的是,联合治疗在这些HCC小鼠模型中显示出明显的抗肿瘤效果(图6G-K),并改变了抗肿瘤免疫的免疫景观,Tregs和MDSCs的比例降低,IFN-γ+和颗粒酶B+ CD8+ T细胞的比例增加(图6I-M)。因此,肿瘤中CD36+ CAFs数量较少可能预示着HCC中更好的免疫治疗应答,并且SSO靶向CD36可以协同提高免疫治疗的疗效。

图6. CD36+ CAFs预测肝癌免疫治疗的疗效,靶向MIF在肝癌小鼠模型中与免疫治疗协同
结论
总之,作者的研究结果在单细胞水平上提供了一个全面的HCC转录组学景观,并确定了由脂质过氧化诱导的CD36+ CAFt分泌的MIF调节肿瘤细胞免疫逃避的新机制。靶向CD36可有效提高免疫治疗的疗效。需要进一步的研究来确定诱导不同CAF亚型形成和激活的肿瘤内信号,并确定其他CAF亚型在TME和肿瘤免疫中的作用。
实验方法
单细胞测序、ELISA、CHIP、qPCR、WB、MIF染色、IHC、肿瘤成球试验、流失细胞术
参考文献
Zhu GQ, Tang Z, Huang R, Qu WF, Fang Y, Yang R, Tao CY, Gao J, Wu XL, Sun HX, Zhou YF, Song SS, Ding ZB, Dai Z, Zhou J, Ye D, Wu DJ, Liu WR, Fan J, Shi YH. CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023 Mar 6;9(1):25. doi: 10.1038/s41421-023-00529-zIF: 33.5 Q1 . PMID: 36878933; PMCID: PMC9988869.