






内皮细胞DGKG促进肝癌血管生成和免疫逃逸
摘要
肝细胞癌(HCC)是世界范围内最常见和最致命的癌症之一。肿瘤微环境(TME)导致HCC患者对当前治疗的反应较差,而肿瘤血管内皮细胞(ECs)是显著促进肿瘤进展的基本TME组分。然而,肿瘤血管内皮细胞在肝癌中的具体功能和机制尚不清楚。本文通过筛选并验证特异性二酰基甘油激酶γ(DGKG)在HCC肿瘤血管内皮细胞中高表达。通过单细胞RNA测序(scRNA-seq)、质谱流式细胞术(CyTOF)以及体外和体内研究来研究内皮DGKG的功能和机制。采用多重免疫组织化学(mIHC)染色和流式细胞术评价TME的变化。在功能上,内皮DGKG促进HCC中的肿瘤血管生成和免疫抑制调节性T(Treg)细胞分化。重要的是,作者发现缺氧诱导因子-1α(HIF-1α)在缺氧条件下通过直接与DGKG的启动子区域结合来激活DGKG的转录。上调的DGKG通过募集泛素特异性肽酶16(USP16)促进锌指E盒结合同源盒2(ZEB2)去泛素化和细胞因子转化生长因子-β1(TGF-β1)正反馈环激活,从而诱导肿瘤血管生成和Treg分化,从而促进HCC进展。重要的是,靶向内皮DGKG增强PD-1和VEGFR-2双重阻断的效率。缺氧诱导的EC特异性DGKG高表达通过ZEB2/TGF-β1轴促进肿瘤血管生成和免疫逃避,提示EC特异性DGKG是HCC的潜在治疗靶点。本文于2023年10月发表在《Journal of Hepatology》IF:25.7期刊上。
技术路线
主要实验结果
1. DGKG在HCC肿瘤血管内皮细胞中高表达并与不良预后相关
作者对HCC肿瘤血管EC进行转录组分析,所述HCC肿瘤血管EC从三对HCC样品的肿瘤和邻近正常组织中分离和鉴定,以鉴定肝癌发生后肿瘤血管EC中的基本分子变化(图1A和1B)。结果显示,DGKG、ZNF192P2和PCDHB18P是HCC肿瘤血管EC中基于倍数变化的三个主要升高的基因。基于差异表达基因(DEGs)的GO和KEGG途径分析也揭示这些DEGs与细胞分化、增殖、代谢和运动途径显著相关(图1C和1D)。qRT-PCR分析进一步揭示DGKG在肿瘤血管EC(CD31+CD45-)中特异性高度表达,但在白细胞(CD31-CD45+)和其它细胞(CD31-CD45-;图1E-1H)中很少表达。此后,作者专注于内皮DGKG的进一步研究。组织微阵列(TMA)的IF染色证明DGKG在肿瘤-EC中特异性高度表达(图1I-1K)。此外,临床病理学数据显示,随着肿瘤进展,内皮DGKG的表达水平显著增加(图1L)。同时,内皮DGKG的高表达增加HCC患者的肿瘤转移发生率并显著降低无病生存期(DFS)和总生存数(OS)(图1M-1O)。总之,这些发现表明DGKG主要在HCC肿瘤血管EC中表达,并且其水平与HCC进展强烈相关。
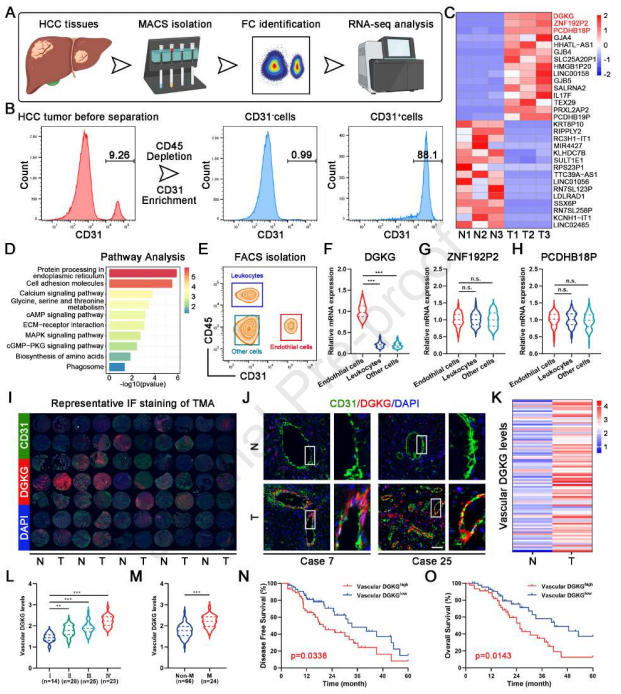
图1 DGKG在HCC肿瘤血管内皮细胞中表达上调,并与不良预后相关
2. 缺氧以HIF-1α依赖方式上调DGKG表达
缺氧通常发生在HCC中,并且与肿瘤进展和不良临床结果密切相关。在缺氧条件下,HIF-1α稳定并促进下游基因的转录。多重免疫组织化学(mIHC)染色显示HIF-1α表达与内皮DGKG正相关,这为缺氧介导的DGKG上调提供证据(图2A和2B)。在此基础上,作者研究HIF-1α在常氧和缺氧条件下对内皮细胞(HUVECs)和肝癌细胞(Huh7)DGKG表达的影响。结果显示,缺氧条件下,HIF-1α过表达和HIF-1α敲低的HUVECs中DGKG的表达水平升高和降低。然而,在Huh7细胞中没有观察到这种现象,表明HIF-1α诱导具有EC特异性的DGKG转录(图2C和2D)。
随后,使用在线工具Jaspar预测DGKG启动子中潜在的HIF-1α结合位点(HBS),揭示4个推定的HBS(图2E)。HBS位点缺失或定点诱变的荧光素酶报告基因测定表明,DGKG启动子中的HBS2和3诱导HIF-1α增强的启动子活性(图2F)。染色质免疫沉淀(ChIP)结果显示,HIF-1α仅被募集到含有HBS2和3的启动子区(图2G)。这些结果表明,HBS2和3对于HIF-1α转录激活DGKG是必需的(图2H)。
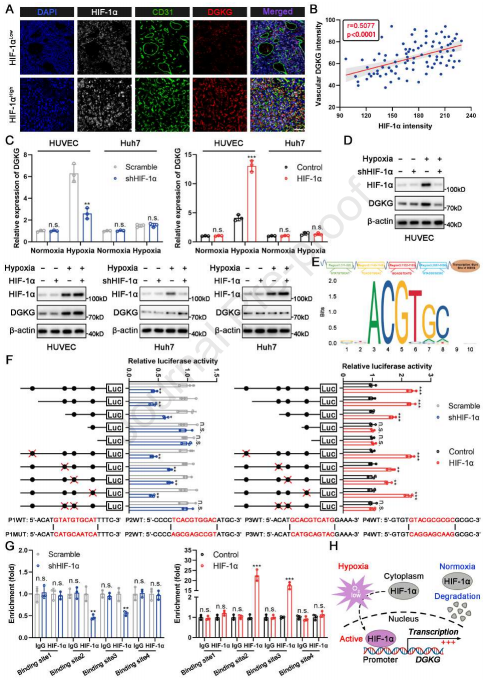
图2 缺氧条件下HIF-1α激活内皮细胞DGKG转录
3. scRNA-seq和CyTOF分析揭示内皮DGKG在HCC进展中的关键作用
作者构建内皮特异性DGKG敲除小鼠和腺相关病毒(AAV)递送的内皮特异性DGKG敲减小鼠,其不改变组织学形态和主要器官中的基础血管稳态。探讨DGKG在肿瘤血管内皮细胞中的作用。首先,作者开发一种肝毒素诱导的肝癌发生模型,通过将二乙基亚硝胺(DEN)与重复CCl4(图3A)组合来描述内皮DGKG在HCC进展中的作用。使用该模型,作者验证DGKG敲除在携带HCC的小鼠中导致较低的肿瘤数量、较小的肿瘤体积和最大化的OS(图3B-3F)。一致地,在HCC的原位移植模型中,作者观察到在注射AAV递送的DGKG敲低的EC的小鼠中对肿瘤进展的类似抑制(图3G-3L)。
为更好地定义肿瘤内皮异质性和DGKG的潜在作用,作者使用10 x Genomics平台对来自DGKGflox/flox和DGKGTek-/-小鼠的HCC肿瘤组织进行scRNA-seq。比较DGKGflox/flox和DGKGTek-/-小鼠之间的DEGs显示,与上皮细胞迁移和粘着斑相关的生物过程和途径在DGKGflox/flox组中富集,表明DGKG是侵袭性肿瘤血管生成所需的(图3M和3N)。由于肿瘤血管内皮细胞对免疫微环境的调节已被广泛报道,作者使用CyTOF分析DGKG在TME重构中的作用。总共70000个免疫细胞,根据它们的标志物表达聚类成10个亚型(图3O和3P)。如图3Q中所示,TGKGTek-/-小鼠中的Tregs显著降低。scRNA-seq和CyTOF数据分析表明,内皮DGKG表达与肿瘤血管生成和免疫逃避之间呈正相关,表明DGKG上调在HCC进展中的作用。

图3 scRNA-seq和CyTOF分析揭示内皮细胞DGKG在HCC进展中的关键双重作用
4. 内皮特异性DGKG缺陷促进血管正常化并促进Treg分化
如上所述,内皮DGKG促进肿瘤血管生成和Treg浸润。因此,为证实DGKG的作用,进行体内研究,其显示在肝癌发生模型中,与DGKGflox/flox小鼠相比,DGKGTek-/-小鼠中CD31标记的肿瘤血管减少(图4A)。同时,在DGKGTek-/-小鼠中沿着肿瘤血管的NG2+周细胞覆盖显著增强,表明内皮DGKG缺失有效地促进肿瘤血管的完整性(图4B)。此外,作者检查内皮DGKG缺失对肿瘤浸润免疫细胞的影响。HCC肿瘤的流式细胞术分析显示,在DGKGTek-/-小鼠中,浸润性T淋巴细胞的存在减少,细胞毒性T淋巴细胞(CTL)的存在增加(图4C和4D)。一致地,AAV递送的特异性DGKG敲低在受损的肿瘤血管生成和Treg支持的免疫抑制方面表现出相似的表型(图4E-4H)。此外,作者将患有HCC的患者分类为低和高内皮DGKG表达组,这表明CD31+血管EC中的DGKG高表达与更致密的血管显著相关(图4I和4J)。此外,mIHC染色的结果显示,与DGKG低表达组相比,DGKG高表达组中Tregs(CD4+和FOXP3+)和CTLs(CD8+和粒酶B+)的积累分别显著增加和减少(图4K-4M)。这些发现有力地表明,内皮DGKG缺陷抑制肿瘤血管生成和Treg分化,促进血管完整性和CTLs扩增,表明内皮DGKG在HCC进展中是必需的。
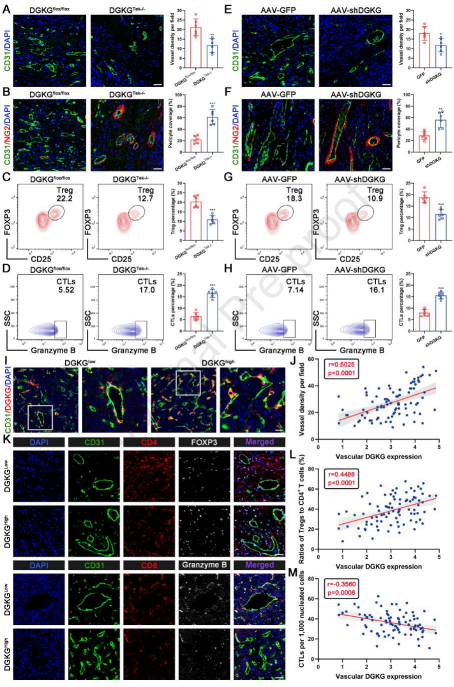
图4 内皮DGKG促进肿瘤血管生成和免疫逃逸
5. ZEB2是DGKG的功能性结合伴侣
为揭示内皮DGKG介导的HCC进展的潜在机制,作者进行免疫沉淀-质谱(IP-MS)分析以探索与DGKG相互作用的蛋白质(独特肽>2),并将其与蛋白质组学(倍数变化>1.5;p<0.05)组合以鉴定与DGKG过表达相关的丰度改变的蛋白质(图5A-5C)。结果显示ZEB2作为与DGKG相互作用并受DGKG调节的潜在靶标(图5D)。ZEB2是上皮-间质转化(EMT)的调节剂,据报道,EMT参与血管生成和免疫应答。因此,作者推测ZEB2可能是DGKG功能所必需的。随后,作者通过外源性免疫共沉淀(Co-IP)、内源性Co-IP和谷胱甘肽S-转移酶(GST)下拉测定验证DGKG和ZEB2之间的直接相互作用(图5E-5G)。进一步的分子作图测定揭示,锌指(ZF)结构域(残基256-429)是DGKG的主要片段,其负责与ZEB2的同源结构域(HD;残基488-703)结合(图5H-5J)。
6. DGKG防止ZEB2泛素化降解
此外,基于蛋白质组学数据,作者研究DGKG对ZEB2表达的影响。蛋白质印迹显示,在DGKG过表达后,ZEB2蛋白表达增加,并且在DGKG敲低后,ZEB2蛋白表达降低(图5K)。另外,DGKG的上调在蛋白质合成抑制剂CHX(图5L和5M)的存在下维持ZEB2的稳定性。蛋白酶体抑制剂MG132而不是氯喹(CQ)逆转DGKG敲减诱导的ZEB2水平的降低,表明DGKG以蛋白酶体介导的方式调节ZEB2稳定性(图5N)。因此,ZEB2的泛素化分别通过DGKG的下调和上调而增强和抑制(图5O)。此外,DGKG抑制ZEB2的K48连接的泛素化(图5P和5Q)。总之,这些结果表明DGKG以泛素依赖的方式稳定ZEB2。

图5 DGKG与ZEB2相互作用并促进其与k48相关的去泛素化
7. DGKG通过招募USP16增强ZEB2去泛素化
考虑到DGKG不能直接影响蛋白质泛素化,作者假设存在一种介导DGKG调控的ZEB2表达的泛素化酶。在IP-MS数据库中,USP16、USP39、TRIM21和NEDD4是负责DGKG介导的ZEB2表达的潜在候选物(图6A)。值得注意的是,仅USP16与DGKG和ZEB2相互作用并抑制ZEB2的K48连接的泛素化和蛋白质降解(图6B-6E)。更重要的是,DGKG特异性增强USP16和ZEB2之间的相互作用(图6F)。GST下拉测定显示DGKG充当促进ZEB2-USP16相互作用的平台(图6G)。此外,DGKG特异性增强由USP16诱导的ZEB2的K48连接的去泛素化(图6H)。
此外,作者模拟DGKG/ZEB2/USP16三联体的3D结构,对接结果显示DGKG的256-429区域中的氨基酸Q359、S360、R375和K376与ZEB2的488-703区域中的氨基酸M571、I572、E573、N574和N576相互作用(图6I和6J)。因此,作者突变DGKG和ZEB2之间的结合位点,所有这些位点都突变为丙氨酸。Co-IP实验表明,DGKG和ZEB2之间的相互作用DGKG(Flag-DGKG-BS-Mut)或ZEB2(HA-ZEB2-BS-Mut)结合位点中的突变所消除(图6K)。此外,当DGKG与ZEB2的结合位点突变时,DGKG在募集USP16与ZEB2结合、减少ZEB2泛素化和增加ZEB2蛋白表达中的作用被消除(图6L和6M)。总的来说,作者认为DGKG可能是一个支架,用于增强ZEB2和USP16的结合,从而形成一个调节复合物,增强ZEB2的去泛素化和稳定化。

图6 DGKG通过增强ZEB2和USP16的相互作用来增强ZEB2的去泛素化
8. 内皮细胞DGKG以TGF-β1依赖的方式通过ZEB2促进肿瘤血管生成和Treg分化
由于细胞因子在肿瘤血管生成和免疫中起着至关重要的作用,作者推断内皮DGKG调节细胞因子的产生,从而重塑TME。为验证这一点,使用人类炎症阵列来检测DGKG过表达后介质释放的变化。结果显示,与对照相比,TGF-β1分泌在DGKG过表达的HUVEC中变化最显著(图7A和7B)。TGF-β1是TME中促进肿瘤血管生成和Treg分化的必需细胞因子,考虑到ZEB参与激活TGF-β1正反馈环以促进TGF-β1产生,作者假设DGKG通过稳定ZEB2来调节TGF-β1分泌。作者对肿瘤切片进行mIHC染色,发现DGKGflox/flox小鼠中ZEB2、TGF-β1和p-SMAD的表达显著高于DGKGTek-/-小鼠(图7C和7D)。
此外,来自HCC患者的TMA的mIHC染色显示内皮DGKG表达与内皮ZEB2表达、TGFβ1表达、血管密度和Tregs百分比正相关(图7E-7H)。关于DGKG功能对TGF-β1的需求,作者在内皮DGKG敲低小鼠中施用r.TGF-β1来过表达TGF-β1(图7I),并证明r.TGF-β1在很大程度上恢复DGKG敲低对肿瘤生长的抑制作用(图7J和7K)。这些结果表明,HCC中内皮DGKG的功能主要取决于ZEB2诱导的TGF-β1过量产生,表明内皮DGKG/ZEB2/TGF-β1轴在HCC的TME中起重要作用(图7L)。
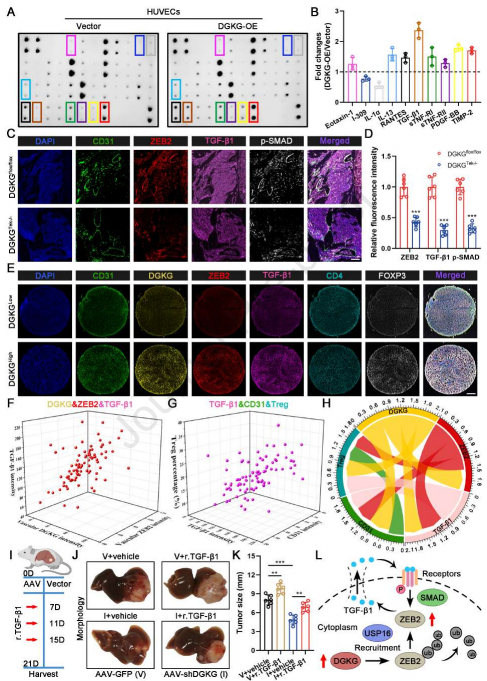
图7 内皮DGKG促进TGF-β1在TME中的表达和分泌
9. 靶向内皮DGKG改善小鼠中PD-1和VEGFR-2双重阻断的结果
最近,靶向TME的治疗策略已成为一种有前途的癌症治疗方法。在HCC小鼠模型中,双重免疫检查点抑制剂(抗PD-1)和抗血管生成剂(抗-VEGFR-2)阻断具有持久的治疗效果并且克服对单独治疗的抗性。由于作者的数据显示内皮DGKG缺陷抑制肿瘤血管生成和Treg分化,基于其有希望的功效,添加抗PD-1和抗VEGFR-2可能提供协同和上级治疗效果。因此,作者在小鼠原位移植肿瘤模型中评估DGKG抑制(AAV-shDGKG)、抗PD-1和抗VEGFR-2(DC101)的组合疗法的功效和效用。治疗3周后,DGKG抑制性小鼠显示出显著延缓的肿瘤生长,并且通过添加抗PD-1和DC101进一步增强这种作用,显示出最强的肿瘤生长抑制和改善的OS(图8A-8D)。同时,在21天的治疗期间,在所有组中没有观察到体重的显著变化,表明这些治疗方案没有严重的不良反应(图8E)。此外,肿瘤切片中的mIHC染色显示,三联治疗具有上级效果,导致肿瘤血管密度显著降低,周细胞覆盖增强,并且Treg积累降低(图8F-8I)。表明三联免疫疗法可能提供更好和更持续的血管控制,并重塑免疫抑制微环境以产生更有效的抗肿瘤应答。
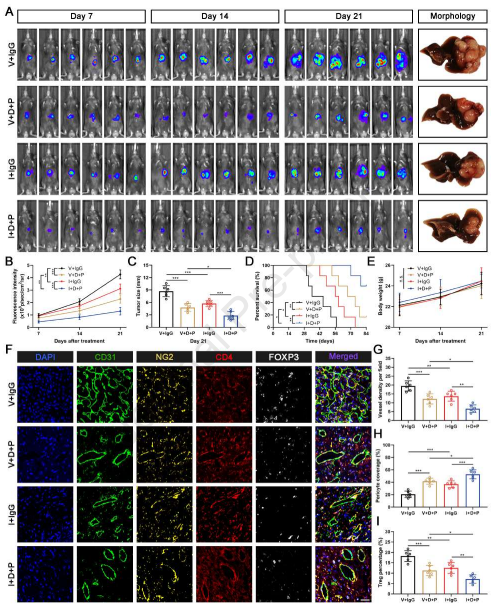
图8 内皮DGKG抑制、免疫检查点抑制剂和抗血管生成药物的三联用有效地抑制HCC的进展。
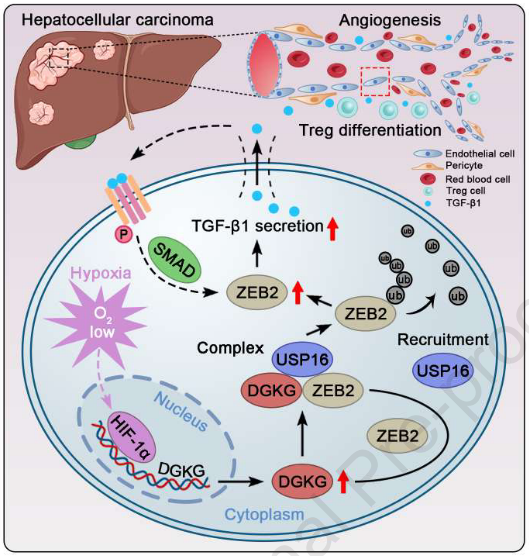
总之,作者的研究结果表明,缺氧诱导内皮细胞特异性DGKG过表达通过募集USP16进行K48连接的去泛素化并诱导随后的ZEB2稳定化,从而增加TGF-β1分泌,从而促进HCC中的血管生成和免疫逃避。最重要的是,内皮DGKG抑制、DC101和抗PD-1的三重组合大大改善DC101和抗PD-1的双重组合在小鼠HCC模型中的功效,显著抑制HCC的恶性进展并改善存活率。这项研究支持靶向内皮DGKG作为精确治疗HCC的潜在策略。
实验方法
scRNA-seq、三维(3D)共培养系统、质谱细胞术(CyTOF)、流式细胞术、Western blotting、多重免疫组化(IHC)测定、免疫荧光染色、qRT-PCR、荧光素酶报告试验、染色质免疫沉淀(ChIP)测定、检测磷脂酸(PA)的产生、无标记蛋白质组学分析、共免疫沉淀、GST-pull down、IP-MS、泛素化试验、分子对接、EdU分析、迁移试验、管形成试验、ELISA。
参考文献
Zhang L, Xu J, Zhou S, Yao F, Zhang R, You W, Yu K, Zhang Y, Baheti T, Pu L, Xu J, Qian X, Xia Y, Dai X, Li Q, Wang X. Endothelial DGKG promotes tumor angiogenesis and immune evasion in hepatocellular carcinoma. J Hepatol. 2023 Oct 12:S0168-8278(23)05170-X. doi: 10.1016/j.jhep.2023.10.006. Epub ahead of print. PMID: 37838036.