






剪接体成分Usp39通过调节自噬参与肝脂质稳态
选择性剪接(AS)的调节使单个转录产物产生多个亚型,从而增加转录组和蛋白质组的多样性。在这里,作者报道剪接体成分Usp39在肝细胞脂质稳态调节中起作用。作者证明Usp39在非酒精性脂肪性肝病(NAFLD)和非酒精性脂肪性肝炎(NASH)受试者的肝组织中表达下调。小鼠肝细胞特异性Usp39缺失导致脂质积累增加,自发脂肪变性和自噬受损。RNA免疫沉淀(RIP-seq)和大量RNA测序(RNA-seq)数据的联合分析显示,Usp39调节了几个自噬相关基因的AS。特别是,Usp39的缺失导致热休克转录因子1(Hsf1)外显子6的5'剪接位点选择,从而导致其表达减少。重要的是,过表达Hsf1可以减轻Usp39缺乏引起的脂质积累。综上所述,作者的研究结果表明,Usp39介导的AS对于维持肝脏的自噬和脂质稳态是必需的。该研究与2023年11月发表于《Nature Communication》上,IF:16.6。
技术路线

主要研究内容
1、NAFLD和NASH患者肝脏Usp39表达降低
识别U4/U6的组件。U5tri-snRNP复合物参与脂肪肝疾病的发展,作者分析了编码16个核心U4/U6的基因mRNA水平。利用GEO数据库(GSE165855)中食粮和HFD喂养的小鼠肝脏转录组学数据,以及GEO数据库(GSE154892)中另一组食粮和CDAHFD喂养的小鼠肝脏转录组学数据研究U5 tri-snRNP成分。其中在HFD和NASH小鼠的RNA-seq数据中,Usp39和Ddx23均显著下调(图1a,b)。接下来,作者对16个U4/U6进行交互网络分析。U5 tri-snRNP成分使用两个RNA-seq数据集,发现Usp39是与NAFLD和NASH小鼠肝脏中U5 snRNP和U4/U6 snRNP成分相互作用最强烈的枢纽因子之一(图1c)。有趣的是,在一个队列(GSE193084)的NAFLD患者中,USP39的表达与NAFLD活动评分呈负相关(图1d)。然后作者将重点放在USP39上进行进一步调查。作者分析了USP39在人类NAFLD患者队列中的表达水平,发现与低NAFLD活动评分组相比,高NAFLD活动评分组USP39明显下调(图1e)。相应地,USP39水平在纤维化晚期(2-4)低于纤维化早期(0-1)(图1f)。在NAFLD和NASH小鼠模型的转录组数据中也检测到USP39表达降低(图1)。接下来,作者通过免疫印迹和qPCR检测了HFD喂养和蛋氨酸胆碱缺乏(MCD)喂养的小鼠肝脏中USP39的表达,并将其与低食喂养的对照组小鼠进行了比较。HFD喂养和MCD喂养的小鼠肝脏中Usp39的表达水平明显低于正常小鼠(图1g、h)。重要的是,研究还发现,与非NASH患者相比,NASH患者肝脏中USP39的表达降低(图1i)。值得注意的是,除了全长波段(65 kDa左右)外,还有一个较低的波段(约55 kDa)(图1g-i)。作者还进行了免疫组化染色,发现Usp39主要定位于细胞核,并且在HFD和MCD喂养的小鼠中,其蛋白水平显著降低(图1j)。总的来说,Usp39在NAFLD和NASH肝脏中的表达降低,暗示了这些疾病的进展中Usp39的作用。
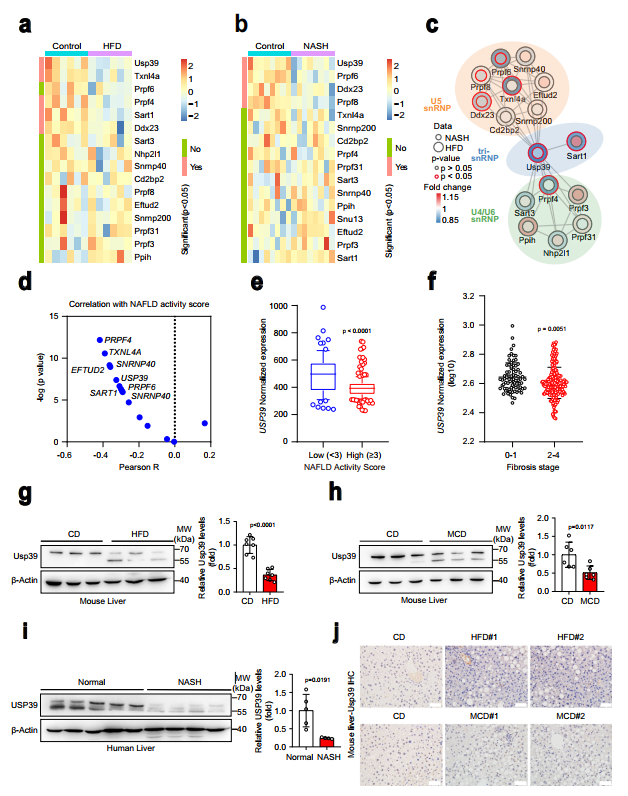
图1NAFLD和NASH患者肝脏Usp39表达降低
2、肝脏Usp39缺失损害肝脏发育和稳态
为探索Usp39在肝脏发育和稳态中的作用,作者通过qPCR和免疫印迹技术检测了Usp39的表达,发现随着年龄的增长,Usp39在小鼠肝脏中的表达水平逐渐降低。然后,作者通过将Usp39fl/fl小鼠与Alb-Cre小鼠杂交,产生肝脏特异性Usp39敲除小鼠(Usp39-HKO)(图2a,b)。Usp39-HKO小鼠在出生后4至8周期间体重显著降低(图2c,d)。在5周龄时,Usp39-HKO小鼠的体重、肝脏体重和肝脏/体重比也有所降低(图2e-g)。H&E染色显示,与对照组相比,Usp39-HKO肝脏中心静脉纤维化,双核细胞数量减少(图h左)。作者还观察到,与对照组相比,Usp39-HKO肝脏中增殖标志物(Ki67和Pcna)肝细胞增殖的表达增加(图2h-j)。值得注意的是,Afp和H19(祖细胞标记物)mRNA表达量显著增加,而Alb蛋白表达量降低(图2k–m)。作者还测量了肝功能的几种标志物(ALT、AST、LDH、白蛋白水平和免疫细胞浸润)。结果显示,在5周龄时,Usp39缺失诱导自发性肝损伤,表现为ALT、AST、LDH水平升高,Alb水平降低(图2n)。通过天狼星红染色和纤维化标志物qPCR分析评估Usp39缺失诱导肝纤维化(图2o,p)。结果显示,这个年龄的Usp39-HKO小鼠未发现凋亡细胞、炎症细胞和祖细胞活化,Usp39缺失在5周龄时诱导自发性肝损伤(图2o,p)。有趣的是,与5周龄Usp39-HKO小鼠相比,10周龄Usp39-HKO小鼠的肝损伤显著减轻(图2n-p)。这些数据提示Usp39在小鼠出生后早期肝脏发育中的重要作用。
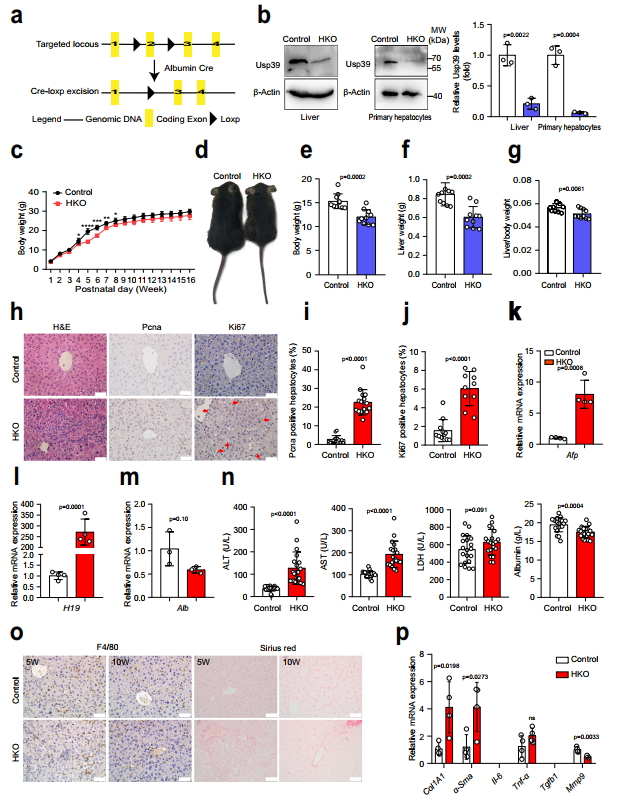
图2 肝脏Usp39基因敲除会损害肝脏稳态和功能
3、肝细胞特异性Usp39缺失诱导自发性脂肪变性
为了确定肝脏Usp39在肝脏稳态和功能中的具体作用,作者在12月龄时检测了Usp39-HKO和对照小鼠。Usp39-HKO小鼠与对照小鼠的体重和肝脏重量均无显著差异(图4a-d)。有趣的是,Usp39-HKO小鼠脾脏增大,肝脏呈浅色(图3a)。H&E染色和Sirius Red染色显示大脂质液滴大量积聚,纤维化(图3b)。与此一致,Usp39-HKO小鼠的肝脏TG水平高于对照组(图3c)。此外,炎症和纤维化基因(Col1a1、α-Sma、Tnf-α和Tgfb1)普遍上调(图3d)。
为了进一步阐明Usp39在肝脂肪变性中的作用,我们用HFD喂养12周的HKO和对照小鼠进行Usp39-刺激。HFD喂养的Usp39-HKO小鼠表现出更明显的肝脏脂肪化和纤维化形成(图3e,f)。肝脏TG水平和炎症和纤维化基因mRNA表达也更显著地增加(图3g,h)。进一步,作者分别通过LC/MS和RNA seq对5周龄Usp-HKO小鼠和对照小鼠的肝脏进行转录组学和脂质组学表征。在肝脏样本中共鉴定出31类脂质中的1692种脂质,其中发现与对照小鼠相比,Usp39-HKO小鼠中TG、二甘油酯(DG)、磷脂酰肌醇(PIP2)和鞘碱(So)显著增加(3i,j)。脂质组和转录组联合分析显示在Usp39-HKO小鼠中肝纤维化和脂肪变性高度富集(图3k-n)。综上所述,这些结果表明Usp39-HKO小鼠更容易患脂肪肝疾病。
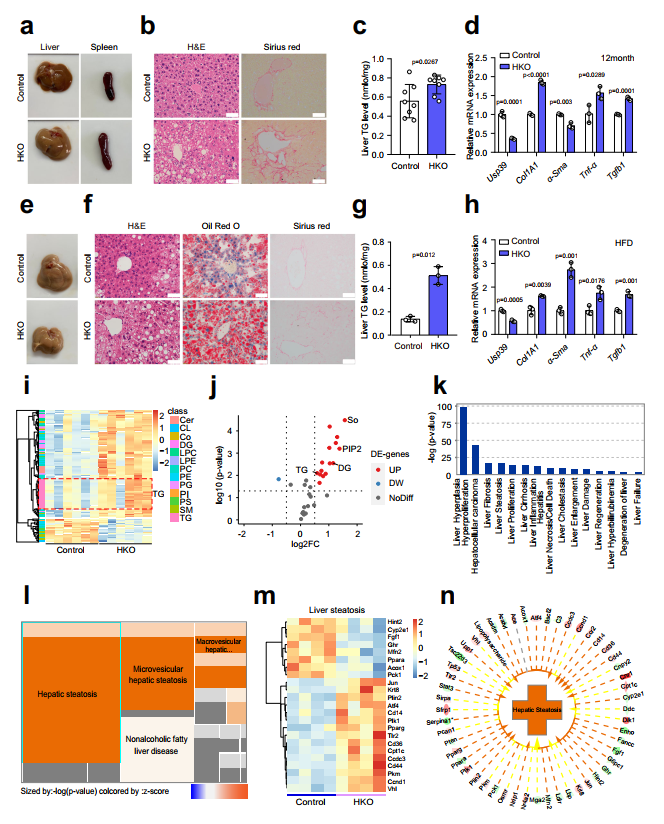
图3 肝细胞特异性Usp39敲除小鼠的自发性脂肪变性
4、肝细胞特异性Usp39缺失小鼠的自噬受到抑制
自噬在肝脏脂滴降解中起着关键作用,作者对脂质组和转录组数据的Ingenuity通路分析显示,与对照组相比,Usp39-HKO肝脏中自噬、TG降解和脂肪酸β-氧化(FAO)通路显著减弱,而mTOR信号通路被激活(图4a)。作者假设Usp39耗竭可能阻断自噬,从而增加肝脏中的脂滴积累。为了探讨这一点,作者通过qPCR评估了自噬相关基因的表达,发现与对照组相比,Usp39-HKO肝脏中的Ulk1、Atg3和Atg7显著下调(图4b)。同样,在Usp39敲低后,AML12细胞(小鼠肝细胞细胞系)和原代肝细胞中Ulk1、Atg3和Atg7的mRNA水平降低(图4c)。此外,免疫印迹显示,敲除Usp39后,Ulk1、Atg7和LC3II降低,p62升高(图4d-f)。作者还通过RT-qPCR检测了p62和LC3BmRNA水平,但未发现对照组和Usp39敲低组之间存在显著差异。在电镜下,Usp39-HKO肝脏中自噬小泡的形成明显减少(图4g)。此外,Plin2和Lamp2的共免疫荧光染色显示Plin2的丰度增加,Lamp2的分布减少(图4h)。同样,在原代肝细胞和AML12细胞中,当Usp39耗尽后,EGFP-LC3点显著减少(图4i)。重要的是,原代细胞和AML12细胞中Usp39的敲除减少了与BODIPYC12染色脂质共定位的EGFP-LC3点的数量(图4j)。这些结果表明Usp39-HKO肝脏中的脂质积累可能是由于自噬受损而发生的。
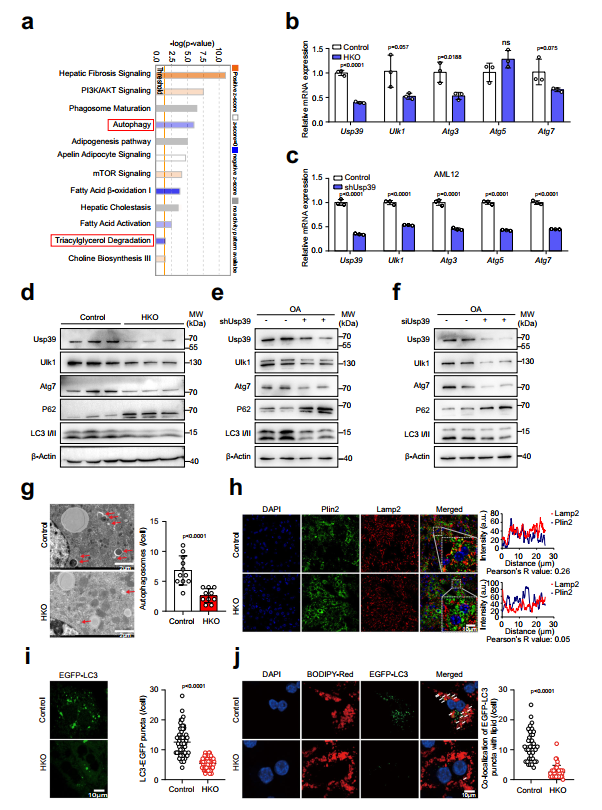
图4缺乏Usp39的肝细胞自噬受损
5、肝细胞特异性Usp39缺失导致自噬相关基因的异常剪接
鉴于Usp39是U4/U6的一个组成部分。U5 tri-snRNP,作者假设Usp39的缺失可能会破坏肝脂质自噬所需的基因的RNA剪接。为此,作者对Usp39-HKO和对照小鼠的肝脏组织(n=4)进行了RNA seq。使用rMATs软件进行AS事件分析,共检测到1472个基因的1993个剪接事件。Usp39缺失的主要AS事件是外显子跳跃(55%),替代5’剪接位点(12%)和内含子保留(12%)(图5a)。热图显示了Usp39-HKO小鼠与对照小鼠相比,肝脏组织中表达的自噬相关基因的差异(图5b),并构建了火山图来显示Usp39缺乏时AS事件的差异分布(图5c)。为了确定Usp39的全基因组结合位点和靶点,作者在AML12细胞中进行了RNA免疫沉淀测序(RIP-seq)。RNA的超声破碎和严格的洗涤条件提高了结合信号的特异性和分辨率。RIP峰分布分析显示,Usp39主要映射到内含子(29%)、外显子(18%)和3'UTR(19%)上(图5d)。对RIP-seq数据进行Motif富集分析,发现GGCCAC和GCCAUA是两个最丰富的元素(图5e)。进一步分析发现,Usp39结合Motif1在自噬基因剪接位点附近明显富集。为了鉴定由Usp39调控的AS候选靶点,作者利用RIP-seq和RNA-seq数据整合了Usp39结合的基因和AS相关基因,鉴定了611个常见基因(图5f)。对611个基因进行基因本体分析发现,与细胞分解代谢过程、剪接体复合体组装、肽基丝氨酸磷酸化和自噬调控相关的通路显著富集(图5g)。为确定由Usp39调控的肝脏脂质积累的功能靶点,作者检查了自噬相关基因的AS模式,这些基因富集于Usp39结合和选择性剪接的转录本中,包括Ulk3、Nrbp2、Tcirg1、Trp53inp1和Hsf1。然后作者通过AS绑定峰和RT-qPCR发现了这些基因的可变剪接。
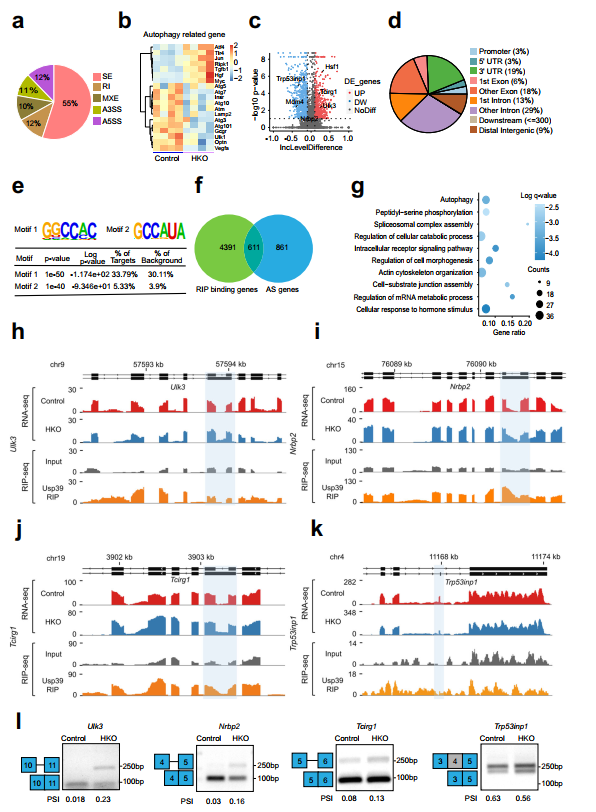
图5 Usp39缺失导致肝细胞中自噬相关基因剪接改变
6、Usp39缺失导致肝细胞中Hsf1的错误剪接和降解
作者下面重点研究了热休克反应的主要转录调节因子Hsf1,它可以促进细胞保护性自噬。作者对Hsf1的剪接事件进行了分析,发现Usp39的缺失导致外显子6的5'剪接位点发生了选择性,引入了一个提前终止密码子(PTC),可能导致无义介导的mRNA衰变(NMD)(图6a,b)。事实上,通过半定量RT-PCR在Usp39-HKO小鼠肝脏中验证了Hsf1的可变剪接(图6c)。小基因剪接实验进一步证实了Hsf1的剪接改变结果随着Usp39添加量的增加而降低,而当Usp39缺失时,Hsf1的剪接改变结果增加(图6d)。随后,作者通过qPCR分析了Hsf1在肝组织中的典型亚型和NMD亚型,发现在Usp39缺失后,典型亚型显著减少,而NMD亚型显著增加(图6e)。与正常喂养的小鼠相比,HFD喂养的和MCD喂养的小鼠在Usp39缺失后,典型亚型也显著减少,而NMD亚型则增加。为了验证NMD亚型通过NMD途径被降解,作者测量了NMD亚型在Upf1敲低和用Actinomycin D处理的AML12细胞中的RNA半衰期,正如预期的那样,Hsf1亚型的NMD半衰期在Upf1敲低细胞中相对于对照组显著增加(图6f)。为了进一步证明Usp39与Hsf1的直接结合,作者在过表达FLAG-Usp39的AML12细胞中进行了RIP实验。RIP-qPCR表明,Usp39结合到Hsf1的外显子6周围的四个位点(图6g),RNA下拉实验进一步显示,生物素标记的Hsf1探针成功地下拉了Usp39(图6h)。因此,与对照组相比,Usp39-HKO肝脏和Usp39敲低的AML12细胞中Hsf1和自噬相关基因的mRNA表达显著降低。相反,在过表达Usp39的AML12细胞中,Hsf1和自噬相关基因的表达显著增加。作者进一步发现,在喂食(图6i)、禁食(图6j)和HFD(图6k)条件下,当Usp39耗尽时,Hsf1、Ulk1、Atg7和LC3II的蛋白水平显著降低,p62的蛋白水平升高。他莫昔芬诱导的Usp39基因敲除小鼠模型(Usp39fl/fl;ugc-creERT2)进一步证实了Usp39缺失后Hsf1蛋白水平的下降。在人细胞系Huh7和HepG2中,Usp39敲低后Hsf1蛋白水平也同样下降。此外,作者观察到HFD喂养的小鼠和MCD喂养的小鼠肝脏中Hsf1蛋白水平较低(图61、m)。最后,与非NASH患者相比,在NASH患者的肝脏中也检测到HSF1表达降低(图6n)。综上所述,这些发现表明Usp39的缺失导致Hsf1剪接改变和快速降解。
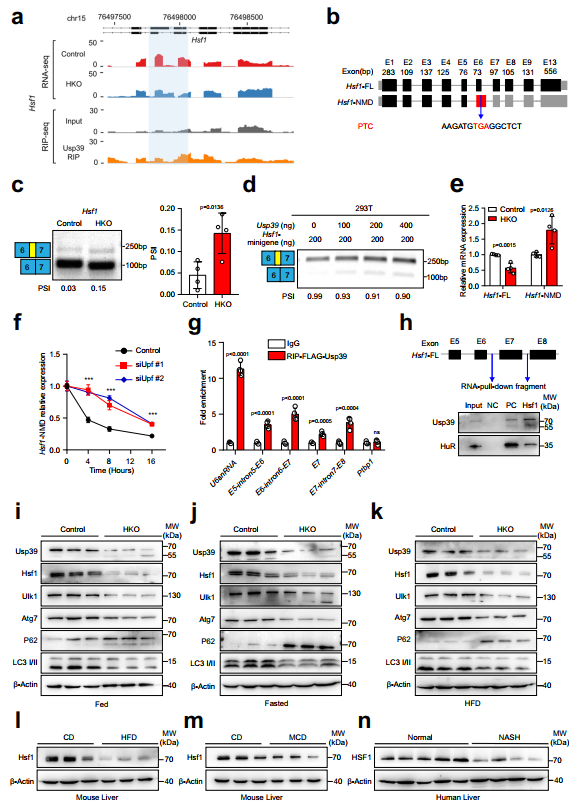
图6 在肝细胞中,Usp39缺失导致Hsf1的错误剪接和快速降解
7、Hsf1促进自噬,减轻Usp39缺失引起的肝脂肪变性
接下来,作者进行了回复实验,以确定Hsf1的下调是否介导了Usp39缺失导致的脂质积累。AML12细胞的BODIPY染色显示,通过强制表达Hsf1可以挽救Usp39缺失引起的脂质积累(图7a,b)。相反,siRNA敲低Hsf1会损害Usp39过表达引发的脂质降解。Plin2和Lamp2染色的免疫荧光进一步显示,在AML12细胞中,通过强制逆转Hsf1的表达,Usp39缺失导致的自噬受损得以恢复(图7c)。相反,在Usp39过表达的AML12细胞中沉默Hsf1会产生相反的效果。免疫印迹显示,在AML12细胞中,特别是在血清饥饿情况下,强行表达Hsf1可以挽救因Usp39缺失而受损的自噬(图7d)。相反,通过敲低Hsf1可消除过表达Usp39引起的自噬增强。为了加强Hsf1是Usp39的功能靶点并促进肝脏脂质代谢自噬的结论,作者向Usp39 floxed小鼠注射AAV8-TBG-Cre或AAV-TBG-Null,以生成肝脏特异性Usp39-HKO小鼠。虽然AAV-TBG-Cre小鼠的体重、肝脏重量和肝/体重比没有差异,但肝脏苍白,肝脏TG水平显著升高。然后将这些小鼠感染表达Hsf1或空载体的腺病毒,然后进行4周的正常饮食。异位Hsf1表达明显减轻了Usp39缺失引起的肝脂肪变性(图7e)。通过Plin2和Lamp2的免疫染色(图7f)和自噬相关基因的免疫印迹(图7g)可以一致地证明,由Usp39缺失引起的自噬受损得以恢复。总之,这些结果表明,由Usp39缺失引起的肝脂肪变性部分是由Hsf1下调和随后的自噬损伤介导的。
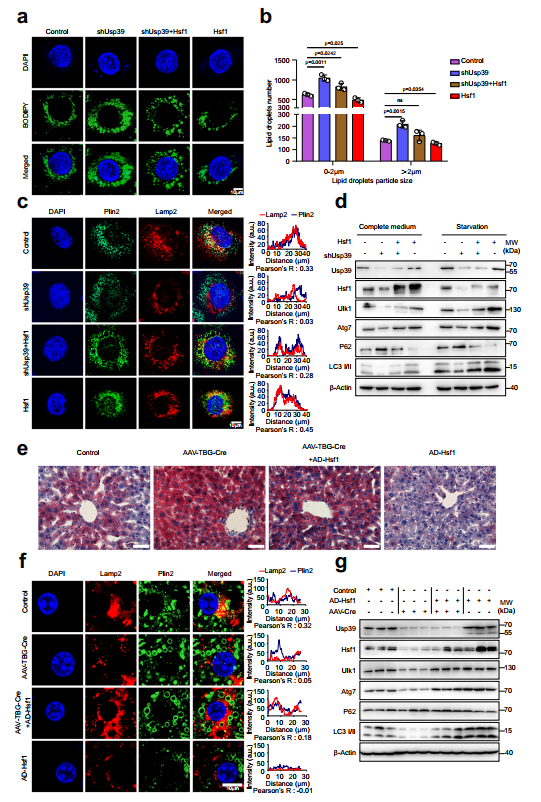
图7 Hsf1促进自噬,减轻Usp39缺失引起的肝脏脂肪变性
结论
该研究结果证明了剪接体成分Usp39在自噬和肝细胞脂质稳态调节中的作用。肝细胞Usp39缺乏导致自噬缺陷和脂质积累,从而导致自发性脂肪变性。在机制上,Usp39调节自噬相关基因的AS,包括Hsf1,以促进自噬。总之,作者的发现AS在调节自噬和肝细胞脂质稳态中的作用使成为治疗脂肪肝疾病的潜在靶点。
实验方法
Usp39fl/+和Albumin-Cre小鼠,小鼠原代肝细胞的分离与培养,组织学分析,免疫荧光染色,体脂和油红O染色,小鼠血清测定及肝功能测定,小鼠代谢评估,透射电子显微镜观察,RNA-seq,RIP-seq,生信分析,线粒体呼吸测量,脂质组学分析,免疫印迹,临床样本分析,RNApulldown
参考文献
Cui D, Wang Z, Dang Q, Wang J, Qin J, Song J, Zhai X, Zhou Y, Zhao L, Lu G, Liu H, Liu G, Liu R, Shao C, Zhang X, Liu Z. Spliceosome component Usp39 contributes to hepatic lipid homeostasis through the regulation of autophagy. Nat Commun. 2023 Nov 3;14(1): 7032. doi: 10.1038/s41467-023-42461-6. PMID: 37923718; PMCID: PMC10624899.