






METTL5通过促进USP5翻译稳定c-Myc来重编程葡萄糖代谢并促进肝细胞癌进展

METTL5是一种18S rRNA甲基转移酶,能够提高癌症细胞中的蛋白质合成活性,对肿瘤发生和细胞命运至关重要。先前的研究表明,缺乏METTL5的小鼠胚胎干细胞(mESC)具有较低的分化潜力和蛋白质合成速率。此外,据报道,METTL5促进乳腺癌和胰腺癌的肿瘤发生,并且METTL5缺陷导致整体蛋白质合成和S6K激活明显降低。尽管已经证明METTL5在调节生物过程中起关键作用,但其在调控HCC复杂代谢重编程中的功能和分子机制仍不完全清楚。因此,为了确定METTL5与葡萄糖代谢的重编程是否存在显著关联,作者联合分析了转录组学和代谢组学的多个层面数据,旨在确定METTL5在HCC中的临床相关性,并阐明METTL5介导的葡萄糖代谢和HCC发展的分子过程。该研究于2023年3月发表在《Cancer Communications》,IF=16.2。
技术路线

主要研究内容
1. METTL5在HCC中上调,与预后不良相关
TCGA-LIHC数据库和GTEx数据库分析显示,METTL5转录水平在HCC中显著上调(图1A)。从GEO数据库下载的微阵列数据同样显示METTL5在HCC中过表达(图1B)。此外,作者发现METTL5转录水平在多种癌症中显著升高,尤其是HCC。根据Kaplan-Meier生存曲线,METTL5高表达的HCC患者预后较差(图1C)。对UALCAN数据库的分析显示,METTL5转录水平与肿瘤分级和分期之间存在显著关联(图1D)。从武汉大学中南医院样本库随机抽取的80对HCC组织及癌旁正常组织中METTL5表达的qRT-PCR分析显示,HCC中METTL5转录本水平显著上调,METTL5表达与肿瘤大小和TNM分期相关(图1E和附表S3)。多因素Cox比例风险回归分析显示,表现为METTL5过表达的HCC患者总生存期较短,METTL5是HCC患者生存期较短的独立危险因素(图1F)。根据中值将HCC患者分为低表达和高表达的METTL5亚组后,Kaplan-Meier分析显示,高METTL5表达与HCC患者的不良预后密切相关(图1K)。Western blotting分析和免疫组化证实了HCC组织中METTL5蛋白水平的上调(图1H-I)。与MIHA人肝细胞的表达相比,5种HCC细胞系中的METTL5 mRNA和蛋白水平增加(图1J-K)。共聚焦激光扫描显微镜发现,HCC细胞中的METTL5蛋白(红色荧光)主要是细胞质(图1L)。这些发现表明,METTL5可能作为诊断和治疗HCC的潜在生物标志物。
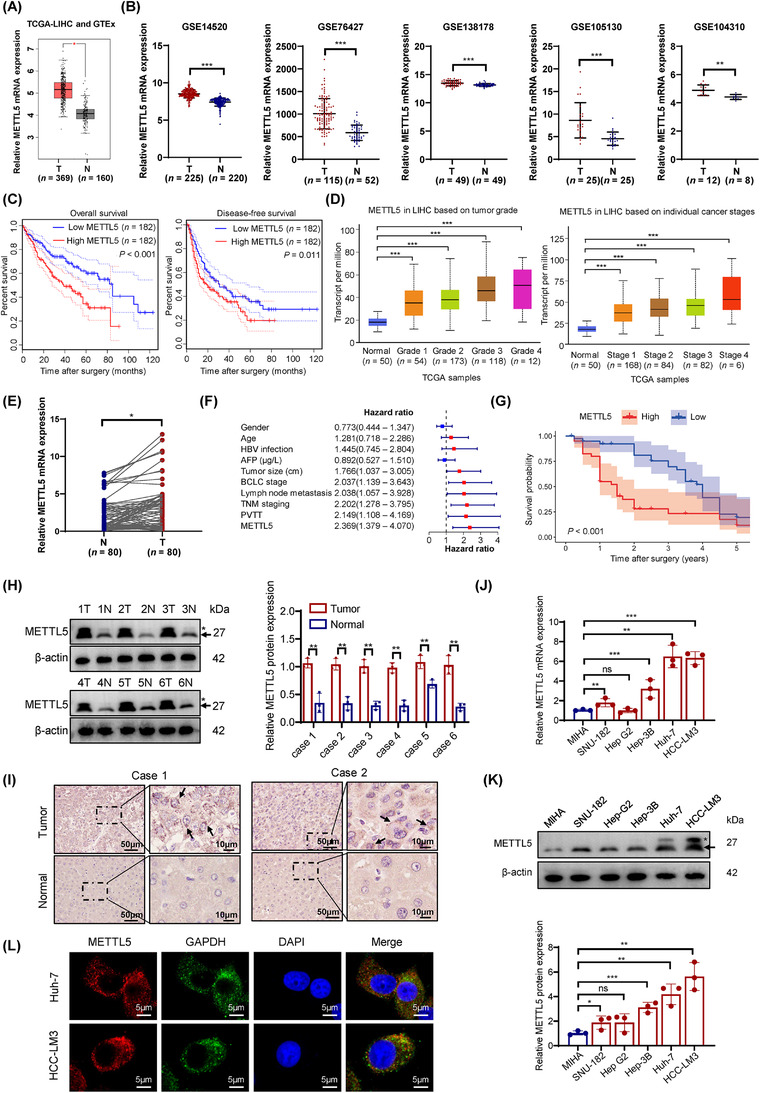
图1 肝细胞癌中METTL5的上调。
2. METTL5增强了HCC细胞中的Warburg效应
作者首先使用CRISPR/Cas9技术构建了稳定的METTL5敲除(KO)HCC细胞系(Huh-7和HCC-LM3),以研究METTL5在HCC中的潜在分子机制(图2A)。然后,作者构建了一个Hep-G2细胞模型,该模型通过慢病毒转导稳定地过表达METTL5。进行RNA-seq以筛选METTL5敲除后在HCC细胞系中差异表达的基因。差异表达mRNA的KEGG通路分析显示糖酵解有相当大的富集(图2B)。对转录组数据进行的GSEA显示,METTL5大大增加了MYC信号通路和糖酵解信号通路。因此,对TCGA数据库的GSEA分析显示METTL5与MYC信号通路之间存在正相关。此外,作者采用基于高效液相色谱-串联质谱的非靶向代谢组学方法检测代谢物。METTL5-KO细胞的代谢组与METTL5-WT细胞的代谢组有显著差异。根据代谢组学分析,METTL5-KO在代谢方面产生了显著的变化,包括更高水平的三羧酸(TCA)循环代谢物和更低的乳酸水平(图2C)。随后,作者在RNA-seq数据中检测到与糖酵解相关的差异表达基因。HCC细胞中METTL5敲除显著抑制糖酵解基因乳酸脱氢酶A(LDHA)、烯醇化酶1(ENO1)、磷酸丙糖异构酶1(TPI1)、溶质载体家族2成员1(SLC2A1)和丙酮酸激酶M2(PKM2)在mRNA和蛋白水平上的表达,而METTL5过表达则发挥相反作用(图2D)。此外,对TCGA-LIHC数据库和80对HCC组织的分析显示,METTL5转录水平与糖酵解基因表达水平之间存在显著的正相关。
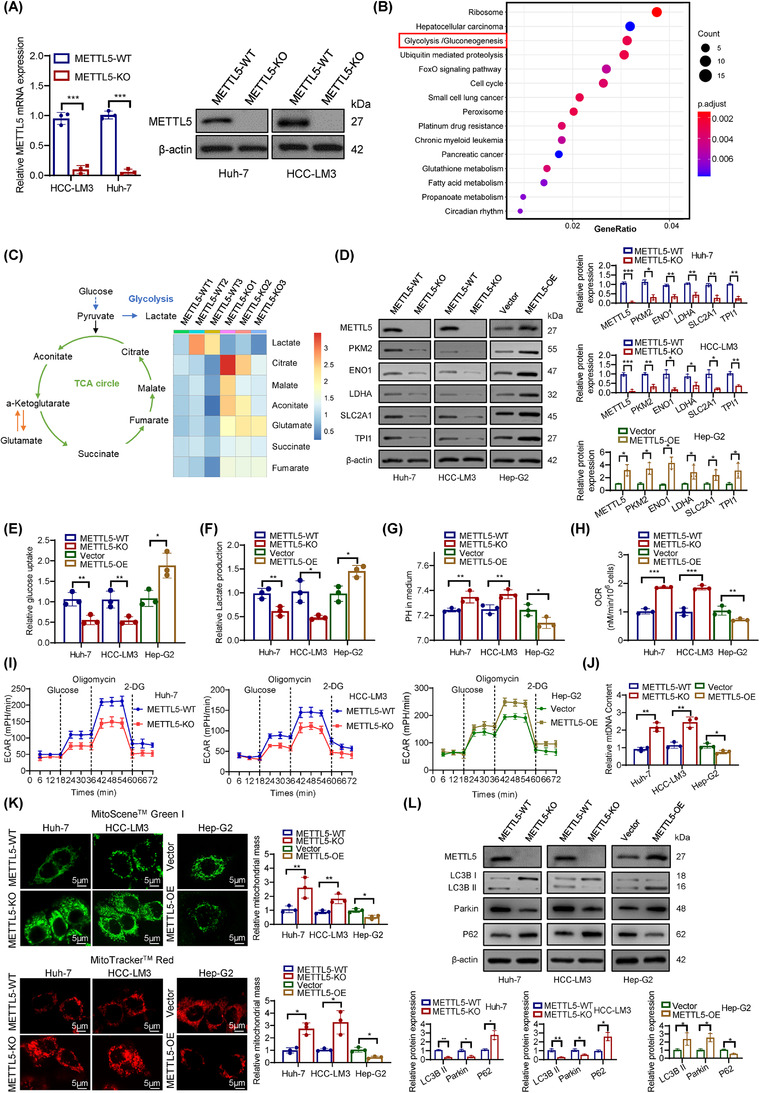
图2 METTL5增强了对HCC细胞的Warburg效应。
随后,作者测量了敲除METTL5后的葡萄糖摄取和乳酸产生,以系统地研究METTL5激活是否促进HCC中的有氧糖酵解(Warburg效应)。这种糖酵解表型被葡萄糖吸收和乳酸生成减少所抑制。正如预期的那样,沉默METTL5显著降低了葡萄糖摄取和乳酸生成。已经观察到酸性肿瘤微环境(较低的pH值)是由乳酸过量产生引起的,这促使肿瘤细胞转移。因此,乳酸合成减少限制了酸性肿瘤微环境的形成,如培养基pH值增加所示。此外,糖酵解基因的下调抑制了肿瘤细胞中线粒体OXPHOS向糖酵解的转化。增强的OXPHOS导致耗氧量增加。结果表明,敲除METTL5抑制了葡萄糖摄取和乳酸生成,增加了HCC细胞的pH值和耗氧量,而METTL5过表达则发挥了相反的作用(图2E-H)。接下来,作者测量了HCC细胞中的ECAR。METTL5敲除显著损害了HCC细胞系的糖酵解,而METTL5过表达则产生了相反的结果(图2I)。作者监测线粒体质量以评估代谢反应;METTL5敲除显著增加HCC细胞中线粒体DNA(mtDNA)含量,而METTL5过表达则发挥相反作用(图2J)。此外,MitoSceneTM系列绿色Ι和Mito Tracker TM系列红染结果显示,METTL5敲除增强了HCC细胞的线粒体功能,而METTL5过表达抑制了线粒体功能(图2K)。据报道,活性氧-氮(RONS)的产生以及缺氧和酸性肿瘤微环境触发线粒体功能障碍并通过Warburg效应增强线粒体自噬。线粒体自噬是一种降解受损线粒体的巨自噬形式,是线粒体对各种应激和代谢紊乱的适应性反应。作者进一步探讨了METTL5是否通过线粒体自噬调节mtDNA和线粒体功能。首先,作者分析了自噬体标志物的蛋白水平,如P62、Parkin、微管相关蛋白1轻链3β-1(LC3B-I)和LC3B-II。P62是一种线粒体自噬接头蛋白,其还原表明线粒体自噬。Western blotting结果显示,METTL5敲除下调了Parkin和LC3B-II的表达,但上调了P62的表达(图2L)。敲除METTL5后,TEM显示自噬体和线粒体减少。根据这些发现,METTL5增加了糖酵解,同时降低了HCC细胞中的线粒体OXPHOS,这与Warburg效应一致。
3. c-Myc在葡萄糖代谢的重编程中起着至关重要的作用
根据之前的一项研究,转录因子在调节糖酵解代谢中起着至关重要的作用。作者假设METTL5通过诱导转录因子的表达来促进糖酵解基因的转录。作者使用UCSC和JASPAR数据库来预测与糖酵解基因LDHA、ENO1、TPI1、SLC2A1和PKM2结合的转录因子。c-Myc、缺氧诱导因子1亚基α(HIF-1α)和MYC相关锌指蛋白(MAZ)可能是这些糖酵解基因的转录因子。结果表明,METTL5敲除降低了c-Myc蛋白的表达,但不影响c-Myc、HIF-1α或MAZmRNA的表达或HIF-1α和MAZ蛋白的表达,这与GSEA结果一致(图3A)。因此,作者怀疑c-Myc可能是将METTL5与糖酵解联系起来的铰链。c-Myc过表达减弱了METTL5敲除介导的糖酵解基因下调(图3B-C)、葡萄糖摄取和乳酸产生下调(图3D-E)、pH值和耗氧量上调(图3F-G)和ECAR的下调(图3H)。此外,c-Myc过表达减弱了METTL5敲除介导的线粒体呼吸和质量增加(图3I-K系列)。这些结果支持了METTL5调节c-Myc表达以调节糖酵解活性的假设。
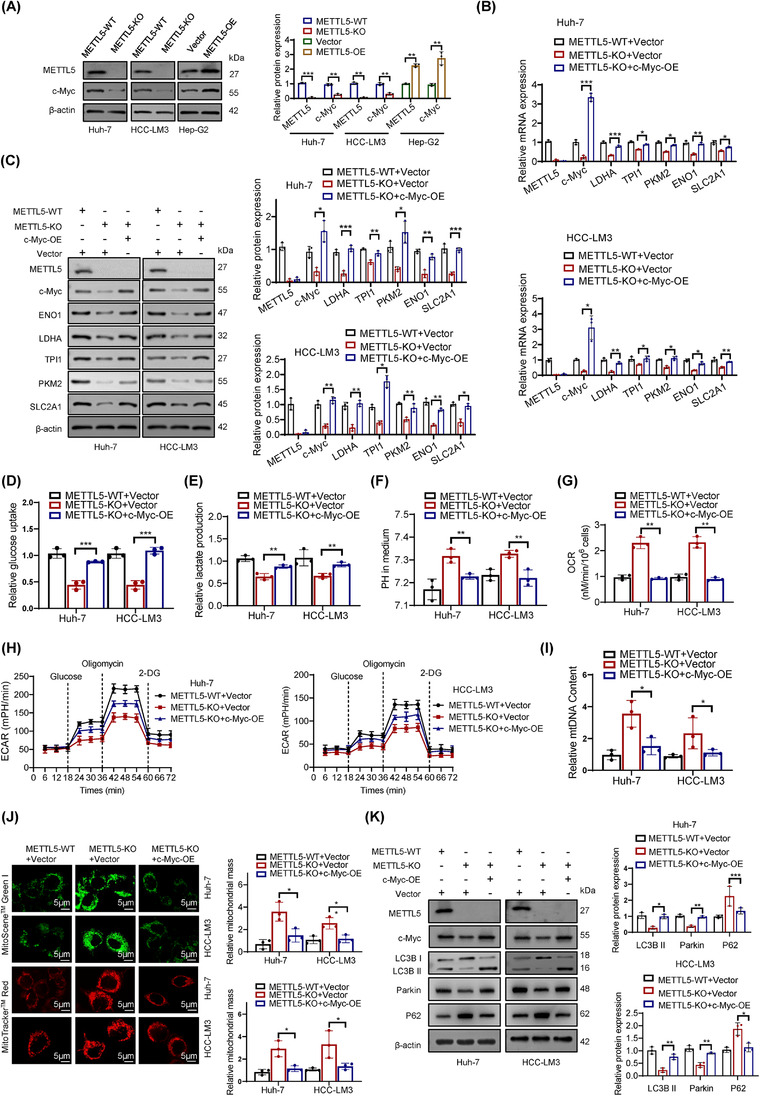
图3 c-Myc在重编程葡萄糖代谢中起着至关重要的作用。
4. METTL5以m6A依赖性方式促进泛素特异性肽酶5(USP5)翻译
根据前期的结果,METTL5敲除显著降低了c-Myc蛋白的表达。此外,多核糖体分离分析表明,在METTL5-KO细胞中,c-Myc的翻译没有显著改变。根据先前的报道,多种泛素结合酶(泛素连接酶)和去泛素化酶通过蛋白酶体系统相互作用并调节c-Myc降解。作者使用翻译抑制剂CHX评估了c-Myc蛋白的稳定性,并证实METTL5延长了c-Myc蛋白的半衰期(图4A)。用蛋白酶体抑制剂MG132进行预处理可防止c-Myc稳定性的提高(图4B),表明METTL5敲除破坏了c-Myc的稳定性,导致其被蛋白酶体降解。随后的泛素化试验表明,METTL5过表达降低了c-Myc的泛素化和降解(图4C)。
作者进行了co-IP和MS鉴定了与HCC中c-Myc相互作用的潜在蛋白质,并研究了METTL5敲除如何促进c-Myc泛素化和降解(图4D)。作者无法鉴定出任何与c-Myc相互作用的METTL5肽。然而,有趣的是,作者发现含有7(FBW7)、F‐box蛋白32(FBXO32)、S期激酶相关蛋白2(SKP2)、USP7、USP37、USP28、USP36和含有32的三联基序(TRIM32)的USP5、F-box和WD重复结构域与c-Myc强烈相互作用。此外,METTL5敲除降低了USP5蛋白的表达,但没有显著改变其他去泛素化和泛素化相关蛋白的表达和翻译水平(图4E)。
METTL5通过甲基化18S rRNA来调节核糖体功能,METTL5-KO细胞的蛋白质比亲本对照细胞少约30%。作者使用METTL5-WT和METTL5-KO细胞的提取物进行多核糖体分离分析,以确定METTL5是否对USP5翻译有影响。结果表明,METTL5敲除对GAPDH mRNA谱没有影响,而USP5 mRNA从较重的多核糖体组分转移到较轻的组分(图4F)。这一结果表明,METTL5敲除抑制了USP5 mRNA的翻译。因此,作者研究了USP5翻译活性是否依赖于METTL5甲基化酶活性。敲除METTL5后,总RNA的m6A水平显著降低,如斑点印迹所示(图4G)。分离rRNA和mRNA后,LC/MS显示METTL5敲除显著降低了18S rRNA的m6A水平,但对mRNA的m6A水平没有影响(图4H)。此外,METTL5-KO细胞和METTL5-WT细胞之间c-Myc的m6A甲基化水平没有差异。然后,作者构建了一个没有酶活性的METTL5突变体。有趣的是,血凝素(HA)-METTL5-WT的过表达减弱了METTL5敲除对USP5的影响,但HA-METTL5-Mut的过表达并不能挽救USP5蛋白的表达(图4I)。因此,USP5的翻译直接依赖于METTL5 18S rRNA甲基转移酶活性。据报道,METTL3-METTL14复合物是一种众所周知的mRNA m6A甲基转移酶复合物,在体外也介导DNA m6A甲基化(m6DA)。然而,没有报告表明METTL5是否参与DNA甲基化。在METTL5-WT和METTL5-KO细胞中,作者检测到基因组DNA的m6DA区域的修饰。作者无法检测到METTL5敲除后的m6DA变化。使用双荧光素酶报告基因测定,作者随后确定METTL5对USP5启动子活性没有影响。结果表明,METTL5是一种纯RNA甲基化酶,对DNA甲基化没有调控作用。
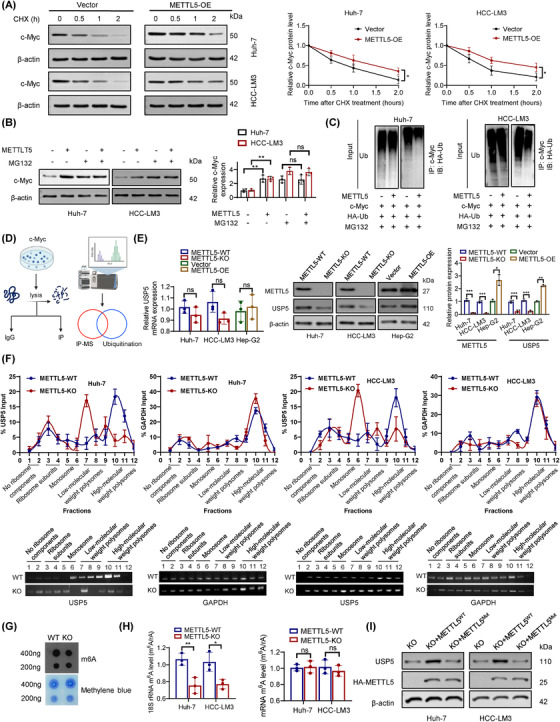
图4 METTL5以m6A依赖性方式促进USP5翻译。
5. METTL5介导的c-Myc稳定需要USP5依赖性去泛素化
作者假设METTL5通过USP5稳定c-Myc。USP5是一种去泛素化酶,对蛋白质的稳定性和功能至关重要。去泛素化酶USP5的底物介导基本的生物学功能,包括细胞存活、增殖和死亡。然而,没有报道描述USP5如何去泛素化c-Myc。USP5敲低显著降低了HCC细胞系中的c-Myc蛋白水平。然后,作者研究了USP5是否通过去泛素化来稳定c-Myc。免疫荧光共定位和核质分离表明,c-Myc定位于核质中,USP5存在于细胞质和核质中(图5A‐B),表明c-Myc和USP5主要共定位在细胞核中。随后,使用内源性和外源性Co-IP测定显示了USP5和c-Myc之间的相互作用(图5C-D)。谷胱甘肽-S-转移酶(GST)下拉试验进一步证实,从细菌中纯化的GST-c-Myc(而非GST)下拉重组His-USP5(图5E)。重要的是,USP5过表达显著提高了c-Myc蛋白的稳定性,同时降低了c-Myc泛素化(图5F-G)。蛋白酶体抑制剂MG132预处理抑制了c-Myc水平的增加。USP5是一种∼120kDa蛋白,具有五个不同的结构域:两个ZnF结构域(1-168;169-289)、一个C盒结构域(290-624)、一个UBA1/UBA2结构域(625-749)和一个H盒结构域(750-835)。为了研究USP5的哪个结构域与c-Myc相互作用,将293T细胞与Flag-USP5和HA-c-Myc的不同结构域共转染,作者的结果表明只有U3和U4直接与c-Myc蛋白结合(图5H)。此外,USP5在K48键处抑制c-Myc多泛素化,但在其他键处不抑制c-Myc多泛素化(图5I)。当K48泛素化位点(K48R)发生突变时,USP5介导的c-Myc泛素化被消除(图5J)。这些结果表明,USP5特异性结合c-Myc并保护c-Myc免受多泛素化介导的降解。最后,在METTL5敲除存在下,USP5的过表达消除了METTL5对c-Myc的影响。这些数据表明,METTL5介导的c-Myc稳定需要USP5依赖性去泛素化。此外,作者使用免疫组化和蛋白质印迹法测定了HCC组织样本中METTL5、c-Myc和USP5蛋白的含量。根据相关性研究,METTL5蛋白水平与USP5和c-Myc蛋白水平呈正相关。与附近的正常肝组织相比,METTL5、USP5和c-Myc在HCC组织中表达强烈。最后,对TCGA-LIHC数据库和20对HCC组织的分析表明,USP5和c-Myc转录水平与糖酵解基因表达水平呈正相关。
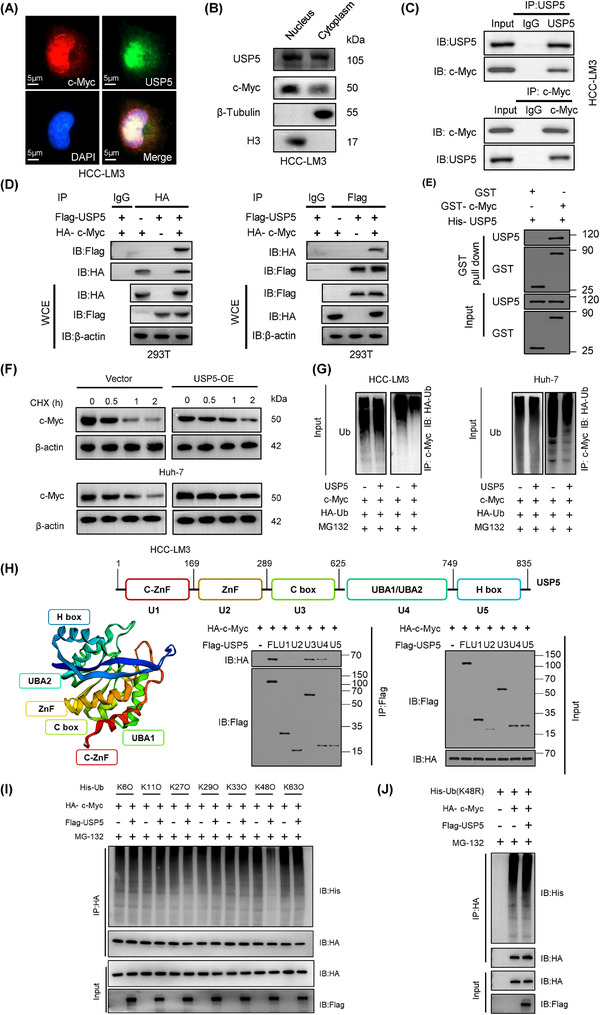
图5 USP5是c-Myc的去泛素化酶。
6. METTL5促进HCC生长和转移
接下来,作者研究了METTL5敲低和过表达对HCC细胞增殖和迁移的影响,以进一步探讨METTL5在HCC中的功能。CCK-8、EdU和集落形成试验显示,METTL5敲除显著降低细胞增殖,METTL5过表达增加增殖(图6A-C)。Transwell和划痕愈合试验显示,METTL5敲除抑制细胞侵袭和迁移,METTL5过表达增加侵袭和迁移(图6D-E)。癌细胞必须经历上皮间充质转化(EMT)才能开始转移。因此,作者研究了METTL5是否对HCC细胞中的EMT有影响。qRT-PCR和Westernblotting结果显示,敲除METTL5下调间充质标志物(N-钙粘蛋白、纤连蛋白和波形蛋白)的表达,但上调E-钙粘蛋白的表达。相反,其过表达显示出相反的效果。这些结果表明METTL5促进了EMT。此外,METTL5缺失大大提高了细胞凋亡率,但对细胞周期没有影响(图6F)。构建HCC皮下异种移植模型、原位HCC小鼠模型、尾静脉注射肺转移模型和腹部转移模型,进一步研究METTL5在体内的作用。METTL5-KO组肿瘤生长和体重均显著低于METTL5-WT组(图6G网络)。免疫组化分析显示,METTL5-KO组Ki-67和糖酵解基因表达低于METTL5-WT组(图6H)。TUNEL标记显示,METTL5-KO组的细胞凋亡率明显高于METTL5-WT组(图6I)。在原位HCC小鼠模型中,METTL5-KO组的肿瘤生长明显慢,糖酵解基因表达比METTL5-WT组低得多(图6J)。肺转移肿瘤模型和腹部肿瘤转移模型显示,敲除METTL5显著降低了转移灶的平均生物发光强度和转移性结节数量(图6K-L)。基于这些结果,METTL5在体外和体内促进了HCC细胞的增殖和侵袭。
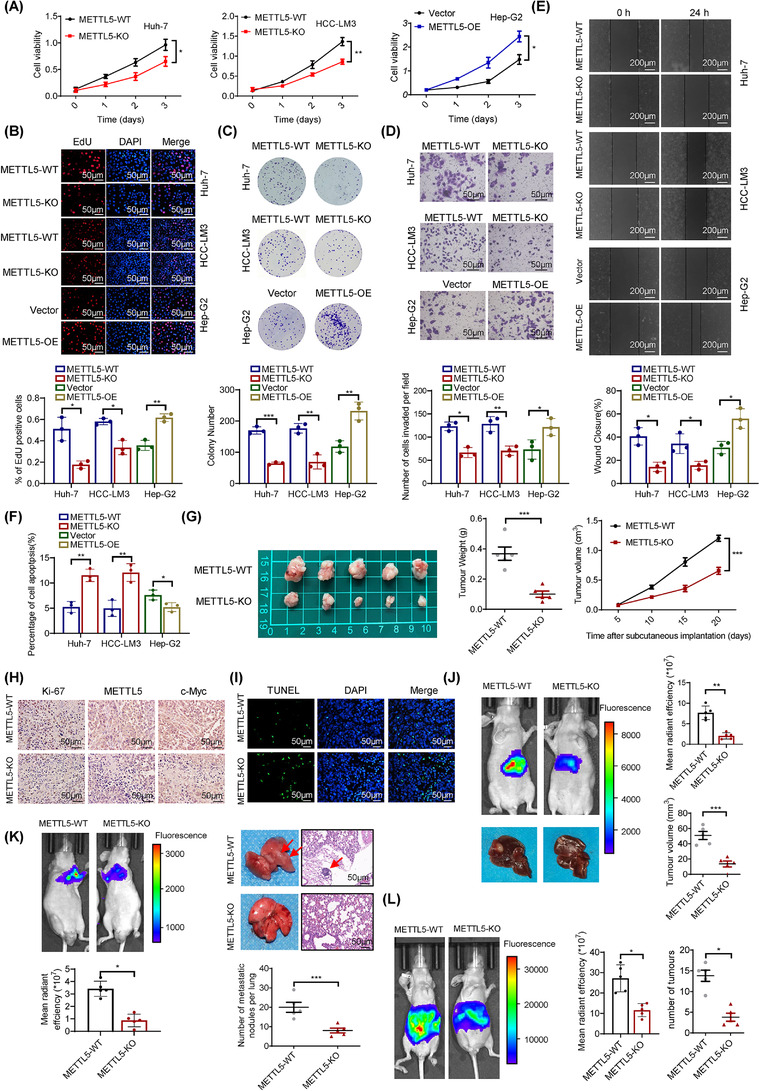
图6 METTL5促进HCC生长和转移。
7. METTL5的肿瘤促进功能取决于c-Myc介导的葡萄糖代谢重编程
由于葡萄糖代谢异常与HCC的进展有关,作者试图进一步确定c-Myc是否是METTL5介导的HCC进展的下游效应子。作者在METTL5-KO细胞系中过表达c-Myc来验证这一假设。一系列表型测试表明,在没有METTL5的情况下,c-Myc过表达降低了对Huh-7和HCC-LM3细胞增殖和侵袭的抑制(图7A-E)。作者进一步验证了c-Myc在裸鼠METTL5介导的HCC进展中的作用,发现METTL5敲除的作用确实被c-Myc过表达逆转(图7F-I)。这些结果表明,c-Myc对METTL5介导的HCC的体外发生和转移具有重要意义。
最后,作者在METTL5-KO细胞系中过表达USP5,以确定USP5的关键作用。一系列表型实验表明,在没有METTL5的情况下,USP5过表达减弱了对Huh-7和HCC-LM3细胞增殖和侵袭的抑制作用。此外,作者在体内验证了USP5参与METTL5介导的HCC进展,发现USP5过表达逆转了METTL5敲低对c-Myc表达和增殖的抑制作用(图7J-K)。这些数据表明,USP5对METTL5介导的HCC癌症促进功能至关重要。
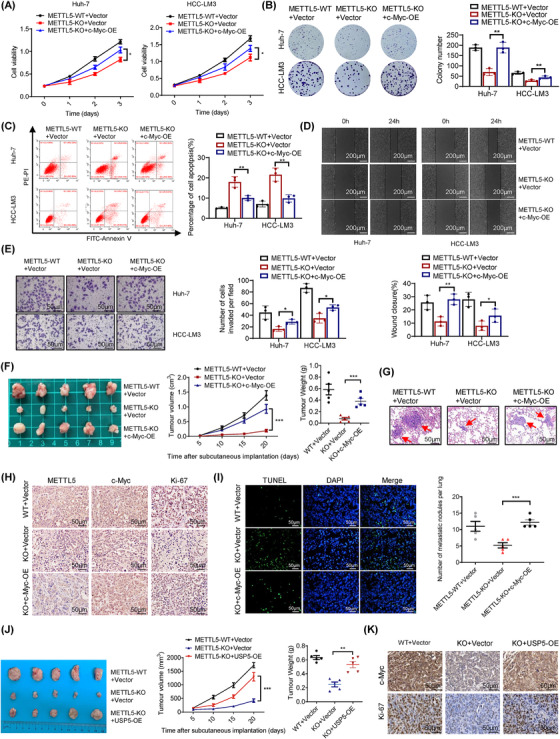
图7 METTL5的肿瘤促进功能取决于c-Myc。
8. cAMP反应性元件结合蛋白1(CREB1)/P300促进METTL5转录
使用GeneCards、UCSC和JASPAR数据库进行计算机模拟研究,以确定与HCC中METTL5上调相关的潜在上游转录因子。CREB1、E74样ETS转录因子1(ELF1)和CCAAT增强子结合蛋白β(CEBPB)是METTL5的主要转录因子,由于CREB1敲低降低了METTL5mRNA和蛋白表达,因此选择CREB1进行进一步研究(图8A-B),但ELF1或CEBPB敲低没有。CREB1转录因子的DNA结合基序是从JASPAR获得的(图8C)。作者生成了一系列截短的METTL5启动子-荧光素酶构建体,以阐明CREB1介导的METTL5转录。荧光素酶报告基因检测结果显示,METTL5基因中碱基对1400至2100的启动子区域包含一个CREB1反应位点(图8C)。因此,作者通过在碱基1554和1565之间顺序突变预测的METTL5启动子序列来构建METTL5-WT和METTL5-Mut启动子荧光素酶报告载体。CREB1过表达增加了METTL5-WT载体的相对荧光素酶活性,但对METTL5-MUT载体的荧光素酶活性影响最小(图8D)。此外,ChIP研究表明METTL5启动子区域与CREB1相互作用(图8E)。如这些结果所示,CREB1通过与METTL5启动子区域结合来增强HCC中METTL5的表达。
此外,作者还研究了CREB1在HCC有氧糖酵解中的关键调控作用。对TCGA-LIHC数据库的分析显示,CREB1转录水平在HCC中显著上调(补充图S19A)。qRT-PCR和Westernblotting分析验证了HCC组织中CREB1mRNA和蛋白水平的上调。此外,TCGA-LIHC数据库的研究表明,CREB1转录水平与糖酵解基因表达水平呈正相关。HCC细胞中CREB1敲除在mRNA和蛋白质水平上显著抑制糖酵解基因LDHA,ENO1,TPI1,SLC2A1和PKM2的表达。TCGA数据库结果与中南医院80对HCC组织的相关性分析显示,METTL5表达与CREB1表达呈正相关。
据报道,E1A结合蛋白p300(p300)作为CREB1转录因子的共激活因子发挥作用,并直接与CREB1相互作用以增强其活性[30].荧光素酶报告基因试验表明,p300可能独立工作以刺激METTL5启动子上的METTL5转录和CREB1活性(图8H)。值得注意的是,当CREB1和p300共表达时,与单独使用基础启动子相比,METTL5转录提高了30倍。这个量明显超过累加量,这意味着CREB1和P300具有功能协同作用。由于METTL5启动子结合位点的突变,这种效应消失了。此外,ChIP分析结果显示p300不能与METTL5启动子区域结合。总的来说,这些结果表明p300在METTL5启动子上作为CREB1共激活因子发挥作用。
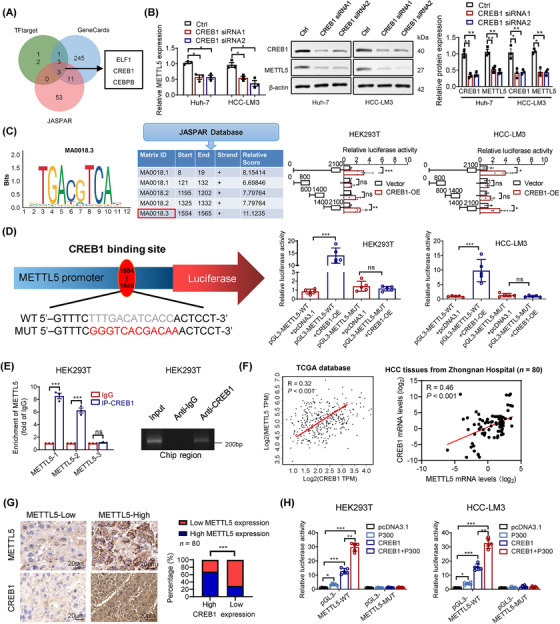
图8 CREB1促进METTL5转录。
9.METTL5敲除抑制PDX模型中的肝肿瘤生长
由于PDX与患者的实际临床标本惊人相似,PDX正迅速成为临床药物研究的黄金标准。将从第二代PDX模型中解剖的肝癌组织植入NCG小鼠体内,以评估METTL5敲除是否对PDX模型产生类似的影响(图9A-B)。腺病毒介导的敲除METTL5具有良好的抗肿瘤作用(图9C-D)。通过蛋白质印迹法证实了METTL5敲除效率(图9E)。
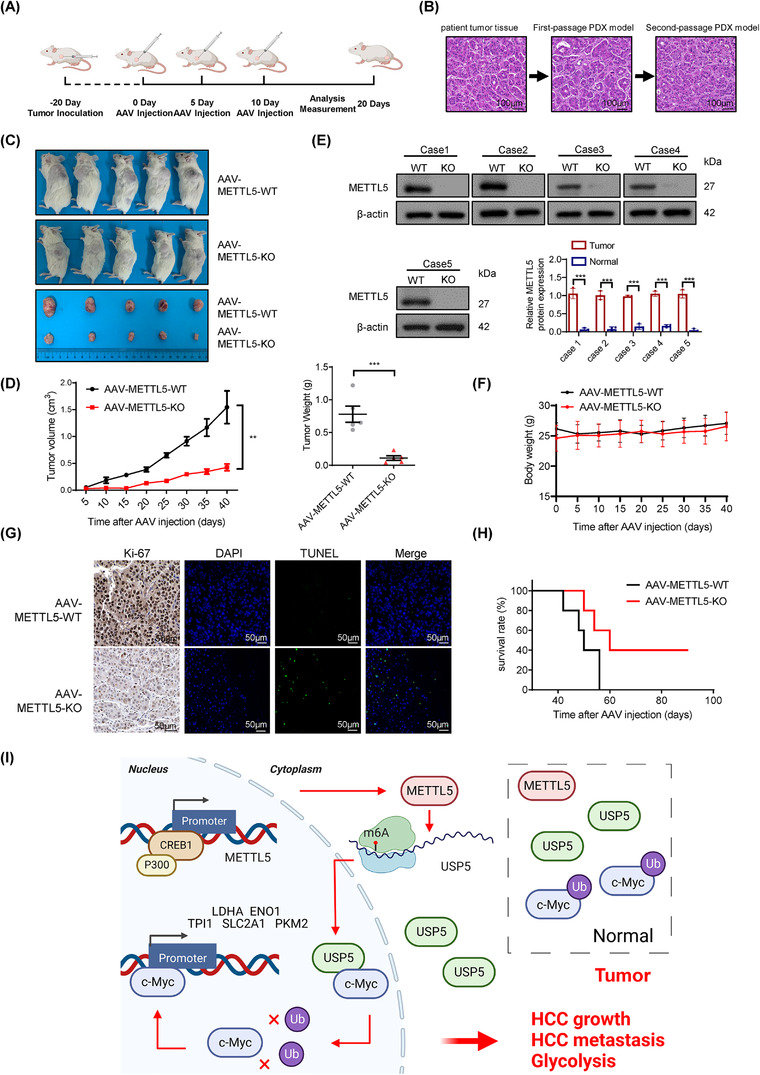
图9 METTL5-KO在HCCPDX模型中的显著抗肿瘤作用。
在治疗期间,所有小鼠的体重保持相似(图9F)。AAV-METTL5-KO组Ki-67表达较低,细胞凋亡率较高(图9G)。METTL5敲除显著延长了生存时间(图9H)。当结合其他结果时,PDX模型中METTL5敲除的显著肿瘤抑制作用为METTL5敲除在临床开发中的巨大潜力提供了强有力的证据。根据集体数据,作者得出结论,METTL5被CREB1/P300激活,通过促进USP5翻译减少c-Myc泛素化,从而导致HCC恶性进展(图9I)。
结语
作者的研究结果为METTL5过表达对HCC的细胞内在影响提供了新的视角,这意味着,除了在RNA甲基化中众所周知的作用外,METTL5还通过许多促癌USP5-c-Myc信号级联调节HCC细胞的增殖和迁移。作者的研究结果为肿瘤代谢相关靶点提供了新的见解,并为未来开发新的HCC治疗方法提供了思路。
实验方法
细胞培养,免疫荧光,免疫组化染色,蛋白质印迹分析,免疫共沉淀(co-IP),泛素化分析,质粒、siRNA、慢病毒构建和细胞转染,蛋白表达和纯化,CCK-8测定,菌落形成测定,划痕伤口愈合运动试验,transwell侵袭试验,细胞凋亡和细胞周期分析,TUNEL染色,小鼠和体内实验,RNA测序(RNA-seq),线粒体形态分析,细胞外酸化率(ECAR)测量,非靶向代谢组学,液相色谱-质谱(LC/MS),双荧光素酶测定,染色质免疫沉淀(ChIP)测定,多核糖体分析,透射电子显微镜(TEM)
参考文献
Xia P, Zhang H, Lu H, Xu K, Jiang X, Jiang Y, Gongye X, Chen Z, Liu J, Chen X, Ma W, Zhang Z, Yuan Y, METTL5 stabilizes c-Myc by facilitating USP5 translation to reprogram glucose metabolism and promote hepatocellular carcinoma progression. Cancer Commun (Lond). 2023; 43(3): 338-364.