






教你四张图搞定一篇nature communication

编码转录因子(TF)基因的致病突变可以影响TF与其同源DNA结合基序的相互作用。TF突变是否以及如何影响与TF复合元件(CE)的结合以及与其他TF的相互作用尚不清楚。在这里,作者报道了B细胞特异性紊乱的人类淋巴瘤中,特别是经典的霍奇金淋巴瘤中TF改变的独特机制。它是由一种复发性体细胞错义突变c.295 T>c(p.Cys99Arg;p.C99R)引起的,该突变靶向干扰素调节因子4(IRF4)的DNA结合域中心,IRF4是免疫细胞中一种关键TF。IRF4-C99R从根本上改变了IRF4-DNA的结合,失去了与经典IRF基序的结合,并获得了与经典和非经典IRF CE结合的新形态。IRF4-C99R通过阻断IRF4依赖性浆细胞诱导彻底改变IRF4功能,并以非经典激活蛋白-1(AP-1)-IRF-CE(AICE)依赖性方式上调疾病特异性基因。这些数据解释了单个突变如何导致TF特异性和基因调控的复杂转换,并为特异性阻断突变TF的新形态DNA结合活性开辟了前景。该研究于2023年11月发表在《nature communication》,IF 16.6分。
技术路线
主要研究结果
1. IRF4-C99R突变在人类淋巴瘤中复发
通过挖掘和整合cHL细胞系的基因组和转录组数据,作者在7个HL细胞系中的2个细胞系,即Bcell衍生的HRS细胞系L428和U-HO1中,鉴定并验证了IRF4基因中相同的c.295 T>c(chr6:394899 T>c;hg38)突变体(图1a)。基于ANNOVAR整合的各种计算机分析(包括SIFT、Polyphen2、MutationTaster、FATHMM、CADD评分),一致预测该变体是有害的。此外,除了在AA100中携带错义突变的单一等位基因(1/251478)外,在影响相邻AA90-104的gnomAD中没有发现种系非同义单核苷酸变体。在HL细胞系中,c.295 T>c突变等位基因伴有至少一个野生型(WT)IRF4基因拷贝,并且WT和突变IRF4 mRNA转录物都同等被检测到(图1a)。由于HRS细胞在受影响的淋巴结中很少见,通过激光显微切割HRS细胞的DNA-PCR验证了IRF4 c.295 T>c在20个原发性cHL样本中的4个样本中存在。IRF4 c.295 T>c最近在原发性纵隔B细胞淋巴瘤(PMBCL)中被描述,该淋巴瘤实体与cHL具有不同的生物学特征。从486例PMBCL病例的不相关队列中平行挖掘靶向基因组测序数据,在486例病例中的29例(5.9%)中发现了相同的IRF4 c.295 T>c突变。相反,IRF4 c.295 T>c在其他淋巴瘤类型中很少被记录,如DLBCL。此外,C99R c.295 T>c(chr6:394899 T>c;hg38)突变的基因组位置在外显子3内,因此位于转录起始位点(TIS)下游>3kb。此外,它缺乏典型的热点RGYW基序,表明这种突变不是由B细胞淋巴瘤的异常体细胞超突变引起的,该突变通常影响TIS下游约2-2.5kb的区域。
IRF4在末端B细胞分化阶段控制浆细胞基因表达程序,尽管在所有亚型中都有高水平的IRF4表达,但在HRS细胞中基本缺乏该程序。在IRF4 c.295 T>c突变中,碱性AA精氨酸取代AA 99位置的中性AA半胱氨酸(Cys;c)(p.Cys99Arg;C99R),其在从人类到斑马鱼的IRF4中高度保守,也在大多数其他IRF家族蛋白的DBD中高度保守(图1a)。C99R位于IRF4 DBD的α3-识别螺旋的中心,并紧邻Arg98,Arg98对特异性IRF4 DNA结合至关重要。这一发现表明C99R可能干扰IRF4:DNA复合物的形成,从而干扰IRF4的转录活性。
2. IRF4-C99R功能丧失ISRE,但被功能激活
为表征IRF4-C99R,首先探索了其与干扰素刺激反应元件(ISRE)的DNA结合活性,该元件包含三个共有基序5’-GAA-3’(图1b),这是IRFs识别的关键基序之一。与IRF4-WT不同,IRF4-C99R不与ISRE结合,如电泳迁移率偏移测定(EMSA)所示。然而,IRF4-C99R突变的复发性和在cHL中的高水平表达表明,这种突变可能不仅构成功能丧失的异常,而且可能具有额外的从头开始的功能。为了分析IRF4-C99R功能,作者产生了四环素(Tet)诱导的表达IRF4-C99和IRF4-WT的BJAB B细胞非霍奇金淋巴瘤细胞的大量培养,其仅在低水平下表达内源性IRF4。时间进程基因表达分析显示,与IRF4-WT相比,IRF4-C99R改变了一组特殊但较少基因的表达(图1c)。这与其失去的结合经典ISRE基序的能力一致。IRF4-C99R与IRF4-WT一样有效地挽救了HRS细胞,使其免于由shRNA介导的内源性IRF4敲除诱导的细胞死亡,从而证实了其功能。
3. IRF4-C99R从根本上改变了IRF4的DNA结合特异性
与在ISRE DNA基序上形成低亲和力同源二聚体或多聚体复合物相反,有效的IRF4 DNA结合需要不同的伴侣,如CEs25-27的ETS和AP-1蛋白。鉴于cHL中普遍不存在ETS TF,作者认为IRF4与HRS细胞中EICE的结合是不可能的。然而,具有高水平JUNB和BATF表达的组成型AP-1活性是HRS细胞的标志。因此,作者推测IRF4-C99R通过DNA结合与最近鉴定的AICE来调节基因表达,间距为4bp 的5’-IRF(TTTC)/nnnn/AP-1(TGASTCA)-3’ (AICE1),或无间距的5’-IFF(GAAA)/AP-1(TGASTCA-3’(AICE2),它们都调节免疫细胞中的关键转录程序。为评估这一假设,作者监测了在强亲和力(标记为“AICE1(Ctla4)”)、弱亲和力(AICE1(IL12Rb))或中间亲和力(AICE2(Bcl11b))AICE的基序条件下IRF4-JUNB/BATF-DNA复合物的形成(图1d)。虽然观察到AICE1(Ctla4)和AICE1的IRF4-C99R结合完全丧失(称为AICE结合模式1(BP1)),但与IRF4-WT(BP2)相比,AICE2(Bcl11b)的IRF4-C99R结合增强(图1d)。IRF4-C99RS104T的行为与IRF4-C992R相似(图1b、d)。引人注目的是,从5’-GAAA-3’到5’-TTTC-3’的AICE2(Bcl11b)中IRF基序的反向互补(称为AICE2FLIP)揭示了仅突变体IRF4-C99R-JUNB/BATF-DNA复合物的形成(图1e,AICE2FLIP,BP3)。此外,AICE复合物的形成通常需要相对于AICE2位于−4 bp(-4T)的胸腺嘧啶(称为AICE2-4T)。IRF4-C99R克服了这种限制,因为它在不存在-4T的情况下形成强的DNA结合复合物,这导致IRF4-WT结合的损失(图1f;AICE2-4C;BP4)。类似地,与IRF4-WT相比,与c-JUN(JUN)/BATF异二聚体AP-1复合物一起观察到IRF4-C99R结合模式的改变。
此外,进行结构建模分析以提供关于IRF4-C99R突变如何影响与ISRE和AICE1 DNA结合基序的相互作用(图1g)。对于结构模型,IRF4和DNA的初始结构是从以前的晶体结构(PDB:7JM4)中获得,并且基于所得的对接参数(如HADDOCK评分、簇大小和去溶剂化能)来考虑最可行的模型。如图1g所示,对于IRF4:ISRE相互作用,用Arg直接取代C99导致与DNA碱基的空间冲突。为了适应C99R和ISRE之间的相互作用,dsDNA必须弯曲和/或扭结。对于IRF4-C99R:AICE1结合,较差的对接得分表明,如较差的能量最小化结构所反映的那样,极不可能发生显著的相互作用。因此,尽管AICE1建模可能由于DNA畸变而受到限制,但它表明IRF4-C99R不与AICE1结合,这与作者的EMSA结果一致。
在仅包含IRF4(AA 20–139)、JUNB(AA 269–329)和BATF(AA 28–87)的DBD重组蛋白中也观察到了IRF4-C99R DNA结合特性的这些改变模式。此外,通过单分子荧光显微镜和交错延时照明观察了IRF4-C99R或IRF4-WT的DNA结合片段, IRF4-C99R的长结合DNA连接(>2s)的百分比与IRF4-WT相当。
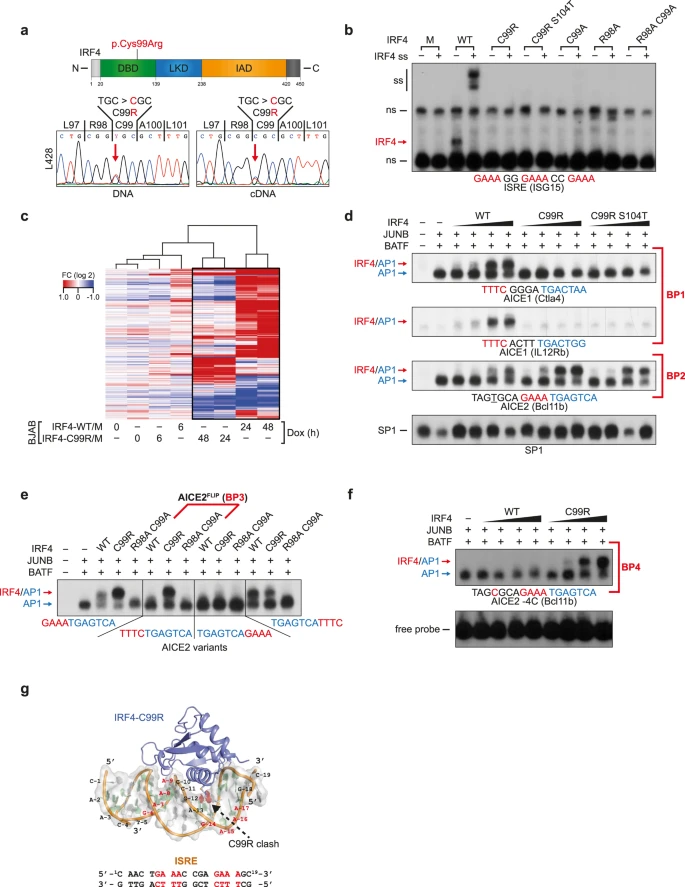
图1:IRF4-C99R功能表征和基本的DNA结合改变
4. IRF4-C99R淋巴瘤细胞中IRF4 DNA结合模式和协同活性发生全局改变
接下来,为详细特异性地绘制HRS细胞中开放性染色质,首先从HRS细胞系L428(携带IRF4-C99R和KM-H2,表达IRF4-WT)以及作为对照的非霍奇金、非IRF4表达REH细胞中生成高分辨率全基因组DNA酶I超敏位点(DHS)和数字足迹数据。DNaseI切割频率的分析揭示了对DNaseI消化的保护作用,表明蛋白质复合物的占据,以及仅在HRS细胞中AICE2(BP2)、AICE2FLIP(BP3)、AICE2−4T和AICE2-4C(BP4)侧翼区域的可及性提高(图2a)。值得注意的是,这些基序在L428IRF4-C99R中富集和保护程度最高(图2a)。这些AICE2基序在L428IRF4-C99R细胞中的共定位分析揭示了与AP-1基序共定位的突变体特异性位点相对应的特异性簇,但与通常参与B细胞和HL细胞基因调控的其他TF位点不一致。这些发现再次支持IRF4-C99R赋予了细胞不同的表达谱的。
为定义L428或KM-H2-特异性DHS组,作者研究了L428IRF4-C99R和KM-H2IRF4-WT细胞之间的标签计数比,并根据它们在DNaseI-seq信号中的倍数变化对它们进行排序(图2b)。L428IRF4-C99R-特异性DHS(即第3组)与这些细胞中上调的基因表达相关(图2b)。然后确定了AICE2(BP2)、AICE2FLIP(BP3)、AP-1和ISRE基序在不同DHS组中的富集情况。L428IRF4-C99R-特异性DHS富集AICE2、AICE2FLIP和AP-1基序,但缺失ISRE基序,而KM-H2IRF4-WT特异性DHs缺失AICE2,AICE2FLIP和AP--1基序,却富集ISRE(图2c)。使用HOMER对细胞系特异性DHS中TF基序的搜索结果显示,AICE2和AICE2FLIP是L428IRF4-C99R-特异性DHSs中富集度最高的两个基序,而不是KM-H2IRF4-WT-特异性DHSs位点(图2d)。相反,ISRE基序在KMH2IRF4-WT-而不是L428IRF4-C99R-特异性DHS中富集,这再次表明IRF4-C9R转移结合位点至AICE2基序(图2d)。L428IRF4-C99R与KM-H2IRF4-WT细胞DHSs的基因集富集分析(GSEA)显示,在上调的基因中,AICE2基序足迹增加,论证了该基序在AICE2 L428IRF4-C99R细胞中的功能相关性。
最后,作者在L428IRF4-C99R和KM-H2IRF4-WT细胞中进行了全基因组范围的JUNB和IRF4 chip-seq分析(图2e-i)。IRF4-JUNB-ChIP峰内的序列紧密聚集(图2e),并且与KM-H2IRF4-WT和GM12878细胞相比,L428IRF4-C99R细胞中的序列显示出更大的重叠(图2f),这与IRF4-C99 R与IRF和AP-1 CE的强制结合一致。尽管与GM12878相比,两个HRS细胞系中的IRF4 ChIP峰值频率更高(图2f),但在KM-H2IRF4-WT细胞中与JUNB的重叠要低得多。当单独排序时,IRF4和JUNB在L428IRF4-C99R中显示出高度相似的结合模式,但在KM-H2IRF4-WT细胞中没有,这对应于开放的染色质区域,并与基因表达增加有关(图2g)。与这些分析和DHS数据集的基序发现结果一致(图2h),在L428IRF4-C99R-特异性IRF4-CIP峰中,AICE2(BP2)和AICE2FLIP(BP3)基序频率增加,相反地,与KM-H2IRF4-WT特异性ChIP峰相比,ISRE基序频率较低。当将L428的IRF4和JUNB染色质结合模式与GM12878细胞进行比较时,也观察到了这些发现。重要的是,GSEA揭示了IRF4和JUNB-CIP峰与L428IRF4-C99R细胞中基因表达增加有关,但与KM-H2IRF4-WT细胞中的基因表达无关。
在ChIP-seq数据集中发现AICE1(BP1)是KM-H2IRF4-WT细胞中最重要的基序,但在L428IRF4-C99R细胞中未被鉴定到(图2i)。AICE2(BP2)出现在两个数据集中最重要的基序中,在L428IRF4-C99R细胞中更为重要,其中共鉴定了五种基序类型(图2i)。分析还揭示了AICE2FLIP(BP3)在L428IRF4-C99R细胞中的独特重要性。这些结果与DNA结合研究一致(图1),并进一步支持IRF4-C99R从根本上改变了淋巴瘤细胞中IRF4全基因组DNA结合模式,并在不同的新AICE上加强与AP-1/JUN TF协同结合。
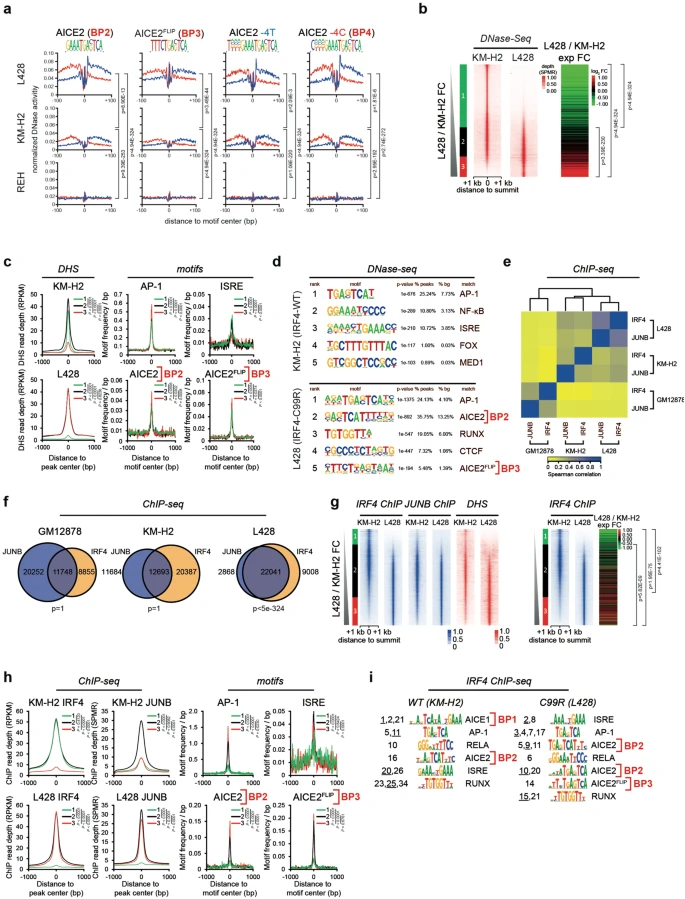
图2:IRF4-C99R与C99R突变阳性淋巴瘤细胞中规范和非规范AICE2位点的全基因组增加和不同的DNA结合模式有关
5. IRF4-C99R破坏原代B细胞中IRF4的功能并重新编程基因表达
为进一步探索IRF4-C99R在B细胞中表达的功能,用IRF4-WT、IRF4-C998R或功能丧失(LOF)突变体IRF4-R98AC99A作为对照,逆转录病毒转导原代小鼠C57BL/6脾脏B细胞(图3a)。在已知的IRF4-R98AC99A LOF突变体中,与IRF4:DNA复合物形成密切相关的残基R98和C99都被丙氨酸(A)取代,从而消除IRF(4)的DNA结合和功能。用LPS和IL−4培养的B细胞导致强大的内源性IRF4表达,并诱导约30%的浆母细胞,其特征是CD138高和B220低表型。在非功能性IRF4-R98AC99A变体异位表达后获得相同的结果(图3a)。IRF4-WT异位表达后,约70%的细胞转化为浆母细胞表型。相反,IRF4-C99R减少了发育中的浆细胞数量,即阻断了固有浆细胞形成,这表明IRF4-C99在B细胞终末分化方面具有主要的负向调节功能(图3a)。为检测基因表达的变化,作者分离了用不同IRF4变体转导的小鼠C57BL/6脾脏B细胞,然后进行RNA-seq分析(图3b–f)。总体而言, IRF4-C99R显示出与R98AC99A LOF变体相比更类似于IRF4-WT的转录谱(图第3b段)。IRF4-C99R调节减少的基因集(图3c),包括IRF4-WT靶基因表达的广泛损失以及新靶点的增加(图3d)。修饰的脾 B 细胞的 mRNA 表达谱与来自各种造血细胞类型的 mRNA 表达谱的集成显示 IRF4-C99R 调节整体 IRF4-WT 诱导和浆细胞特异性基因表达的阻断(图 3e),证实 IRF4-C99R 无法指导 IRF4 定向浆细胞进程。同时,IRF4-C99R 上调髓系相关基因(图 3e、f)。总之,这些数据证实了与IRF4-WT相比,IRF4-C99R依赖性基因调控和功能的根本变化。
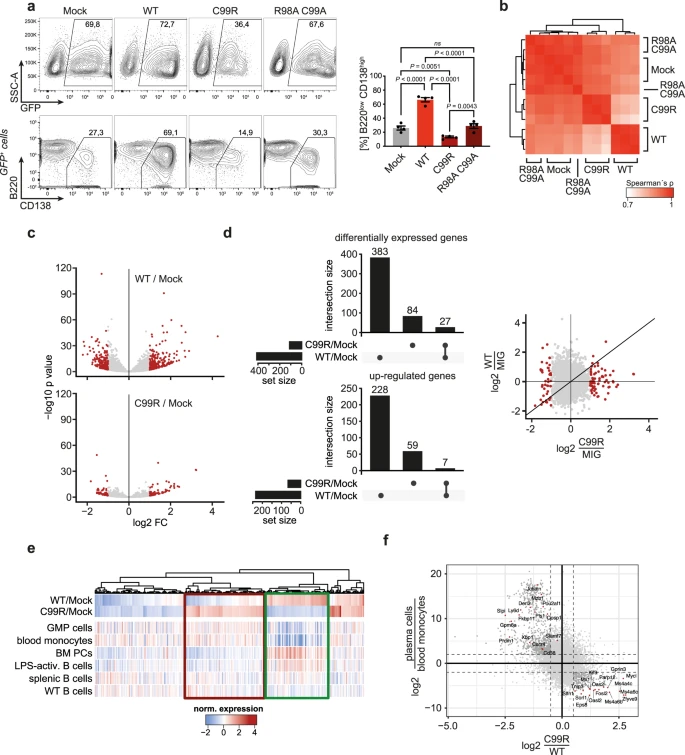
图3:与IRF4-WT相比,IRF4-C99R阻断IRF4依赖性浆细胞诱导并调节数量较少但不同的基因
6. IRF4-C99R通过非经典AICE激活淋巴瘤特异性基因表达
为将IRF4-C99R调节的基因与cHL的HRS细胞特异性固有基因直接联系起来,作者整合了来自脾脏B细胞的RNA-seq数据与HRS细胞特异性基因表达谱(图4a)。在 IRF4-C99R 仅上调而非 IRF4-WT 上调的 HRS 细胞特异性表达基因中,GATA3、CCL5(也称为 RANTES)和 TNFRSF8 (CD30),这三种基因都是最突出的 cHL 标志性基因,以及 CD80、PDE4D 和 CASP6(图 4a)。
为了进一步剖析这些基因的IRF4-C99R特异性诱导机制,重新分析了L428IRF4-C99R细胞特异性IRF4-JUNB ChIP峰的ChIP-Seq数据,但在KMH2IRF4-WT细胞中未发现。作者专注于调控GATA3表达的区域,在L428IRF4-C99R特异性IRF4-JUNB ChIP峰中鉴定了几个AICE2样CE,分别为GATA3Peak_1(5’-TGAGTCAGAGA-3’)、GATA3Peak_2(5’-TAAATGAGTCA-3’)和GATA3Peak_3(5’-GGAATGAGTCA-3’)(图4b,左)。DNA结合研究表明,IRF4-C99R在这些位点形成IRF-AP-1复合复合物,而IRF4-WT不与这些串行结合(图4b,右,AP-1由JUNB/BATF异二聚体组成)。值得注意的是,这些位点均不包含经典的5’-GAAA-3’IRF基序,而是包含其非经典简并变体。这些结果表明,与C99相比,IRF4 R99在这些基序上的灵活性更高,类似于对图1e-g中描述的AICE2变体的观察。与KM-H2IRF4-WT细胞相比,在使用ExplaiNN的ChIP-seq数据中鉴定的含有IRF基序在L428IRF4-C99R细胞中退化得更多,这反映了IRF4-C99 R对退化的半含ISRE基序的结合能力的增加(图 4c),这在 AICE 基序中最为明显(图 4c)。通过分析荧光素酶报告基因构建体证实了 IRF4-C99R 介导的 GATA3Peak_1 组件的转录活性(图 4d)。在这里,IRF4-C99R 与 AP-1 TFs JUNB 和 BATF 联合使用特异性增强了荧光素酶活性,而 IRF4-WT 则没有。最后,携带IRF4-C99R突变的HRS细胞系与缺乏IRF4-C99 R的HRS的表达谱与cHL特异性基因关系的比较表明,cHL标志性基因GATA3、CCL5和TNFRSF8的表达在IRF4-C99R细胞系中特别高(图4e)。
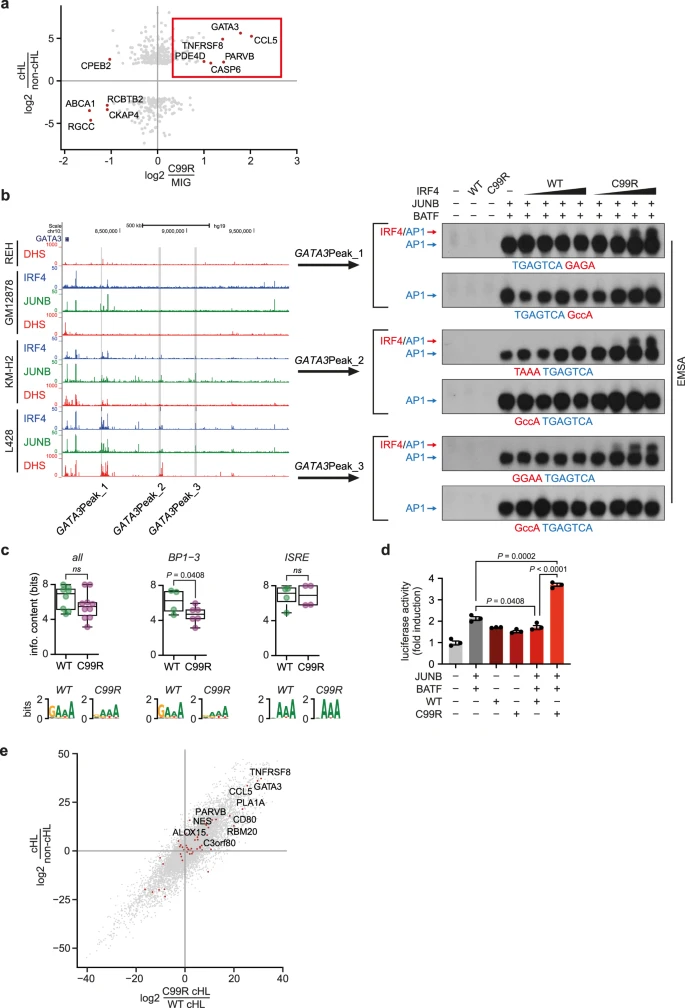
图4:IRF4-C99R以非经典AICE2依赖的方式上调包含cHL标志性的基因
结论
在这里,作者描述了人类淋巴瘤,特别是cHL中TF改变的独特机制,涉及靶向IRF4的DNA结合域中心的复发性体细胞错义突变c.295 T>c(p.Cys99Arg;p.C99R)。他们发现,IRF4-C99R导致IRF4的DNA结合特性发生根本性变化,结合到经典IRF基序的结合损失和结合到经典和非经典IRF CE的新形态获得。在功能上,证明IRF4-C99R阻断了IRF4依赖性浆细胞诱导,并以非经典激活蛋白-1(AP-1)-IRF-CE(AICE)依赖的方式上调疾病特异性基因。
实验方法
细胞培养,细胞转染,电泳迁移率变化分析(EMSA),免疫印迹,桑格测序,PCR,免疫组化,重组蛋白的克隆与纯化,ChIP-seq,DNase-seq,RNA-seq,流式细胞术
参考文献
Schleussner N, Cauchy P, Franke V, Giefing M, Fornes O, Vankadari N, Assi SA, Costanza M, Weniger MA, Akalin A, Anagnostopoulos I, Bukur T, Casarotto MG, Damm F, Daumke O, Edginton-White B, Gebhardt JCM, Grau M, Grunwald S, Hansmann ML, Hartmann S, Huber L, Kärgel E, Lusatis S, Noerenberg D, Obier N, Pannicke U, Fischer A, Reisser A, Rosenwald A, Schwarz K, Sundararaj S, Weilemann A, Winkler W, Xu W, Lenz G, Rajewsky K, Wasserman WW, Cockerill PN, Scheidereit C, Siebert R, Küppers R, Grosschedl R, Janz M, Bonifer C, Mathas S. Transcriptional reprogramming by mutated IRF4 in lymphoma. Nat Commun. 2023 Nov 7;14(1):6947. doi: 10.1038/s41467-023-41954-8. PMID: 37935654; PMCID: PMC10630337.