






EGFR!ALKBH5!防止胶质母细胞瘤中的铁死亡!
生长因子受体是最重要的致癌途径之一,但药物抑制剂作为单一疗法的效果有限。在这里,作者发现表皮生长因子受体(EGFR)信号传导抑制胶质母细胞瘤干细胞(GSCs)中的N6-甲基腺苷(m6A)水平,而遗传或药理学EGFR靶向升高m6A水平。激活的EGFR诱导非受体酪氨酸激酶SRC磷酸化m6A去甲基化酶,AlkB同源物5(ALKBH5),从而抑制染色体维持1(CRM1)介导的ALKBH5核输出,从而允许细胞核中持续的mRNA m6A去甲基化。ALKBH5通过m6A调节和YTH N6 -甲基腺苷RNA结合蛋白(YTHDF2)介导的谷氨酸-半胱氨酸连接酶修饰子亚基(GCLM)的衰变,精密调节铁死亡。ALKBH5的药理学靶向增强了EGFR和GCLM抑制因子的抗肿瘤功效,支持EGFR-ALKBH5-GCLM的致癌轴。总的来说,EGFR通过ALKBH5去甲基化酶的核保留对表转录组景观进行重编程,以防止铁死亡,为致命癌症的治疗提供了治疗范例。本文于2023年11月发表于《Molecular cell》, IF: 16.0,Q1。
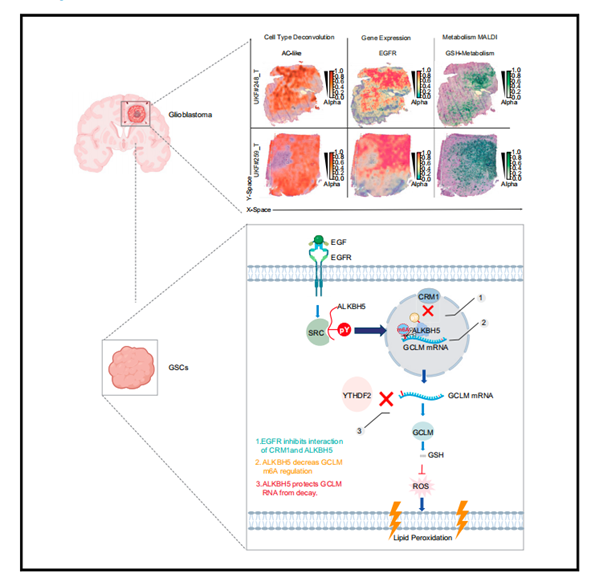 作用机制图解
作用机制图解
技术路线:
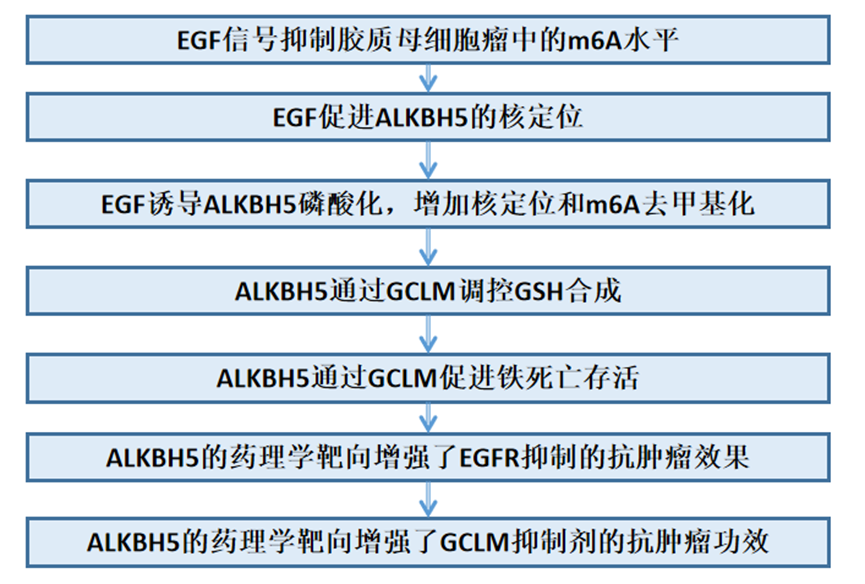
主要实验结果:
1、EGF信号抑制胶质母细胞瘤中的m6A水平
作者最近报道了m6A写入者和擦除者产生的基因表达特征与GSCs中选择的RTK通路(特别是PDGFR、VEGFR和EGFR)相关。PDGFR通过转录诱导METTL3增加m6A水平,而VEGF不改变m6A水平,提示RTKs在表转录组学中具有不同的机制。EGFR和PDGFR是胶质母细胞瘤中两个重要的致癌基因,它们与不同的转录亚型有关,但两者在肿瘤内经常发生改变,显示出肿瘤内的空间异质性。利用最近报道的胶质母细胞瘤的空间多组学分析提供了肿瘤组织学和细胞类型分布(图1A)。胶质母细胞瘤中GFR的空间表达分布显示EGFR和PDGFRA是互斥的(图1B)。EGFR与径向胶质细胞壁龛和星形细胞样(AC样)亚型相关,而PDGFR与少突细胞祖细胞样(OPC样)壁龛和神经祖细胞样(NPC样)亚型相关(图1C、1D)。基于这一背景,作者假设EGFR通过与PDGFR不同的机制调控m6A水平。EGFR在胶质母细胞瘤中稳定m6A解读子YTHDF2,但在肝细胞癌中,YTHDF2通过破坏EGFR mRNA的稳定性来降低肿瘤生长。然而,EGFR对全球m6A水平的调控尚不清楚。作者创建了国外m6A调节器签名,包括写入器(METTL3、METTL14、METTL16、VIRMA、RBM15、RBM15B、ZC3H13和WTAP)、擦除器(FTO和ALKBH5)和读取器/调制器(YTHDF1、YTHDF2、YTHDF3、YTHDC1、YTHDC2、HNRNPC、IGF2BP3、CBLL1和HNRNPA2B1)。
为了直接研究EGFR在m6A调节中的作用,作者询问了患者来源的GSCs的潜在功能关系。由于GSCs通常与EGF一起培养,作者将EGF从培养基中去除1周以避免伪像,然后用EGF配体处理两个患者来源的间充质GSCs,以确定急性EGF配体处理对m6A水平的影响。选择间充质GSCs是因为它们被认为更具侵袭性。使用点印迹法、比色法和免疫荧光法测量,EGF处理逐渐降低了m6A水平(图1E-1G)。由于抗m6A抗体不完全具有特异性,作者采用液相色谱-质谱(LC-MS)并行分析,证实EGF处理降低了GSCs中m6A的表达(图1H和1I)。在相互功能丧失研究中,作者用两种临床使用的EGFR抑制剂(厄洛替尼和拉帕替尼)治疗GSCs。在没有配体的情况下,EGFR可以在胶质母细胞瘤中被激活,包括通过组成型活性变体EGFRvIII的表达。总的来说,这些数据表明,与PDGFR诱导m6A相反,激活的EGF信号会下调GSCs中的m6A。
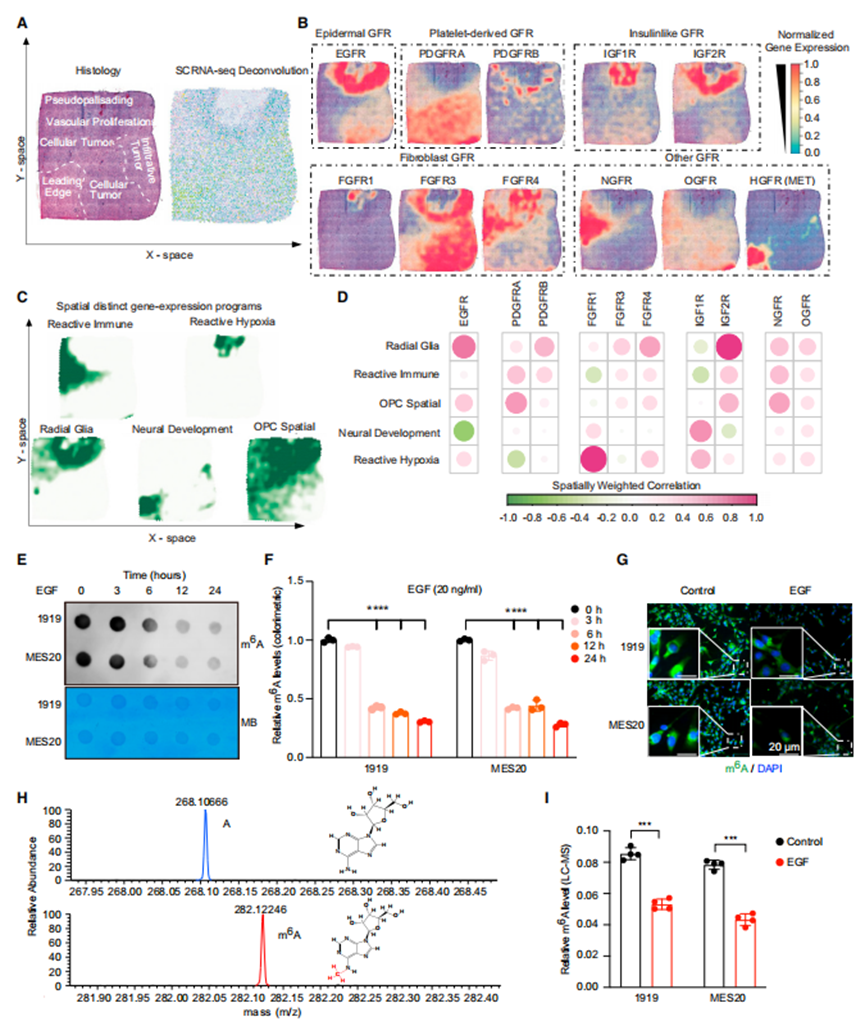 EGFR信号调控胶质母细胞瘤中RNA m6A水平
EGFR信号调控胶质母细胞瘤中RNA m6A水平
2、EGF促进ALKBH5的核定位
为了研究EGF信号调控m6A的机制,作者测量了EGF处理后GSCs中m6A写入和擦除的总蛋白表达。EGF处理没有改变GSCs中m6A调节因子的总蛋白水平(图2A)。同样,使用EGFR抑制剂厄洛替尼和拉帕替尼治疗对m6A调节因子的蛋白水平没有影响(图2B)。由于m6A去甲基化酶FTO的细胞内分布决定了FTO与不同RNA底物的结合,作者测试了EGF是否改变了m6A调节因子的定位。EGF并未实质性改变GSCs中FTO或甲基转移酶复合物(METTL3、METTL14和WTAP)的差异定位(图2C)。相比之下,EGF处理诱导ALKBH5从细胞质转移到细胞核(图2C和2D)。在互失功能的研究中,用短发夹RNA(shRNA)靶向GSCs中的EGFR表达抑制ALKBH5的核定位(图2E和2F)。在平行药理学研究中,作者用EGFR抑制剂厄洛替尼处理GSCs,增加了细胞质ALKBH5的定位(图2G)。组成活性EGFR EGFRvIII的表达促进了ALKBH5在GSCs中的核定位(图2H)。其他ErbB家族成员(ERBB2/3/4)不直接结合EGF配体,但激活类似的细胞内通路,并可与EGFR形成异源二聚体。
接下来,作者推断EGF诱导的ALKBH5核积聚在GSCs中可能是由于核输入增加或向细胞质输出减少。因此,作者测量了EGF处理对ALKBH5结合输入蛋白(核转运蛋白亚基α2, KPNA2;核转运蛋白亚基β1, KPNB1)和输出蛋白(染色体维持1 [CRM1])的影响。总的来说,EGFR活性促进ALKBH5从细胞核输出减少,其功能是去除mRNA m6A修饰。
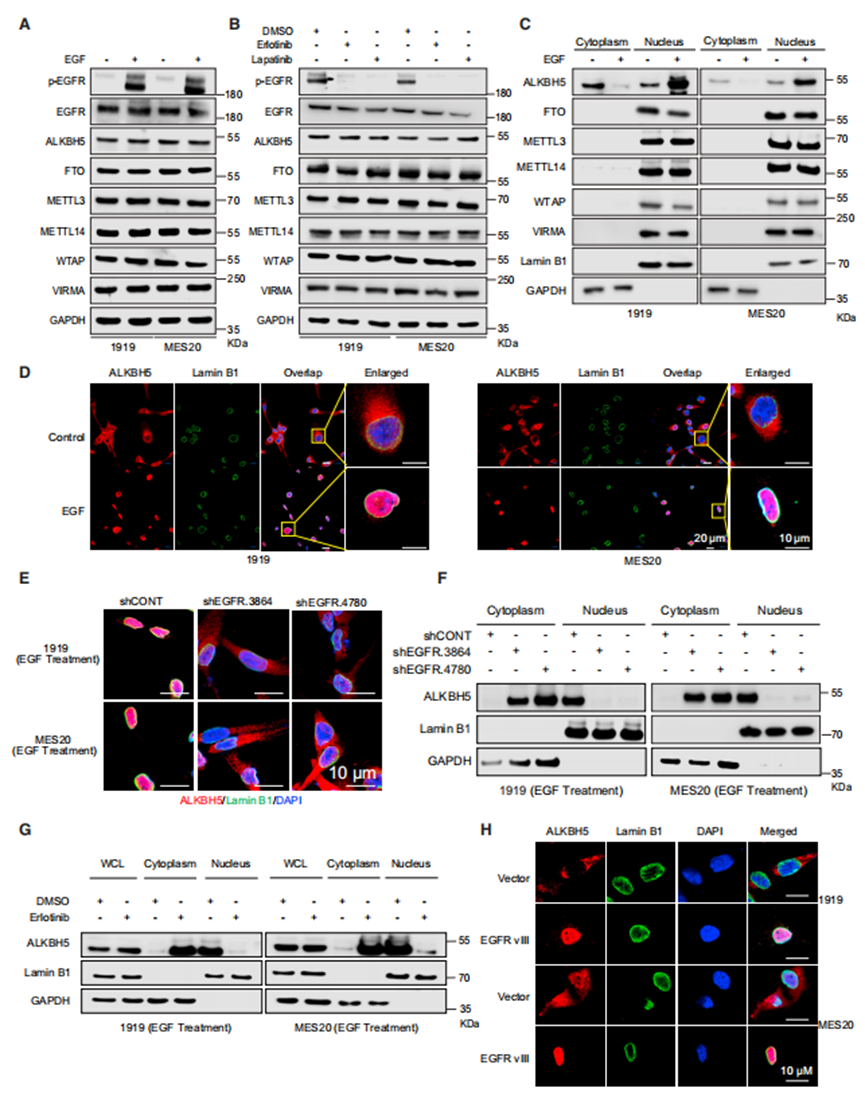 EGF-EGFR信号调控ALKBH5的核定位
EGF-EGFR信号调控ALKBH5的核定位
3、EGF诱导ALKBH5磷酸化,增加核定位和m6A去甲基化
ERK磷酸化METTL3以调节其功能。由于蛋白磷酸化可以调节许多分子的细胞内定位,作者接下来测试了EGFR是否通过ALKBH5磷酸化调节m6A水平和ALKBH5的细胞内定位。EGF处理诱导了GSCs中ALKBH5酪氨酸磷酸化,但没有丝氨酸或苏氨酸磷酸化(图3A)。与EGF配体处理的作用平行,EGFR和EGFRvIII过表达诱导ALKBH5磷酸化(图3B和3C)。反过来,EGFR抑制剂降低了GSCs中ALKBH5的磷酸化(图3D)。
为了确定对EGF诱导的ALKBH5磷酸化重要的特定氨基酸残基,作者使用GPS 5.0预测了ALKBH5蛋白内潜在的磷酸化位点,这表明71号氨基酸的酪氨酸是EGFR和SRC磷酸化的最高可能性位点(图3E)。EGFR增加了野生型(WT) ALKBH5WT和ALKBH5Y306F的磷酸化,但没有增加突变的ALKBH5Y71F的磷酸化(图3F)。接下来,作者研究了ALKBH5Y71对ALKBH5细胞内定位和去甲基化酶活性的作用。用shRNA诱导GSCs减少内源性ALKBH5,然后用耐shRNA的ALKBH5进行转导,EGF处理促进了ALKBH5WT的核定位,而不是ALKBH5Y71F的核定位(图3G)。因此,Y71位点ALKBH5的磷酸化对于ALKBH5的核定位至关重要。
接下来,作者测试了ALKBH5磷酸化对m6A修饰和肿瘤细胞生长的影响。ALKBH5缺失的GSCs显示出大量的m6A水平,而ALKBH5WT显著降低了m6A水平,而ALKBH5Y71F则没有(图3H和3I)。m6A水平的调节反映在肿瘤细胞活力和体内肿瘤生长上。靶向ALKBH5的表达降低了体外细胞活力和体内肿瘤生长,通过重新表达ALKBH5WT而不是ALKBH5Y71F完全恢复了细胞活力(图J - 3L)。总之,作者的数据表明,EGF诱导ALKBH5 Y71磷酸化,这对于ALKBH5核输出和肿瘤细胞活力和体内生长至关重要。
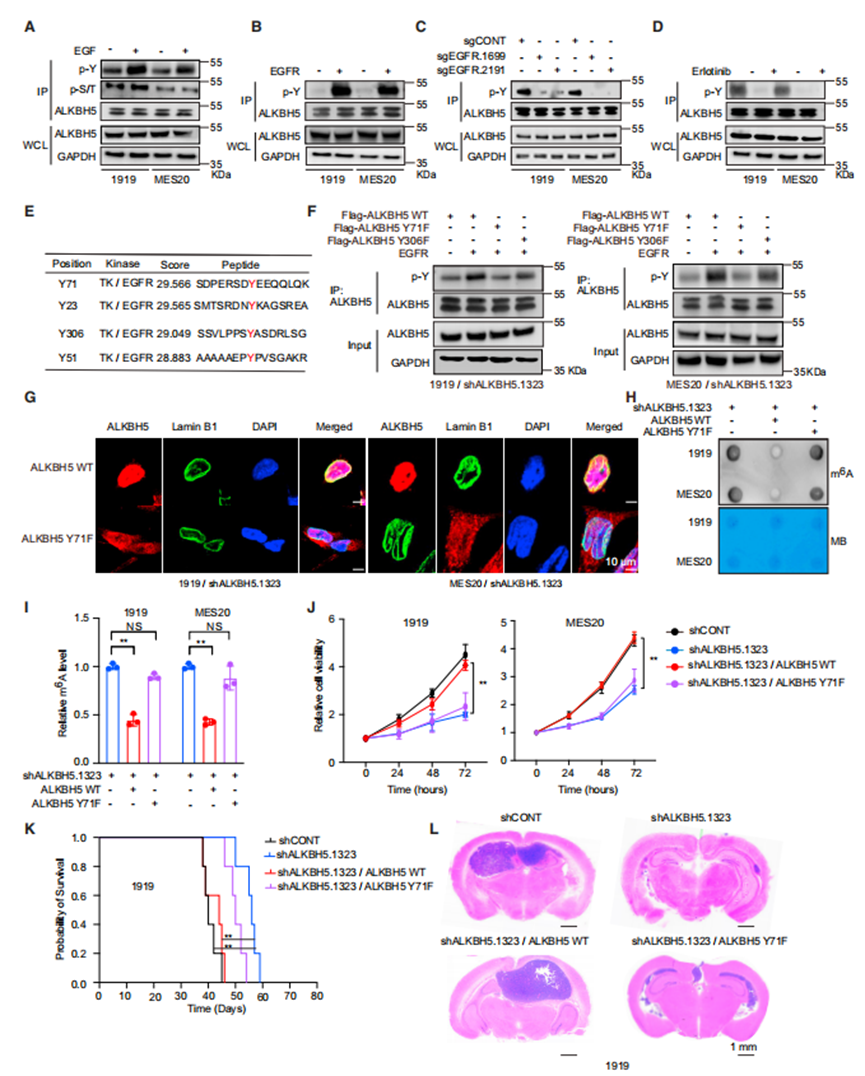 ALKBH5 Y71的磷酸化是其核定位及其在体内和体外功能的必要条件
ALKBH5 Y71的磷酸化是其核定位及其在体内和体外功能的必要条件
4、ALKBH5通过GCLM调控GSH合成
由于ALKBH5对GSC至关重要,作者寻找其作用的下游介质。Venn图显示了一个由17个基因组成的基因簇,其表达与ALKBH5相关,带有m6A修饰,并且在GSCs和患者肿瘤中优先表达(图4A)。在这些基因中,作者之前报道了GSCs对JMJD6和HEXB的依赖性。GSCs表达的GCLM蛋白水平高于NSCs(图4B)、ACs(图4C)和分化的胶质母细胞瘤细胞(DGCs,图4D)。作者探讨了该轴是否调节GSCs的干性。研究表明EGFR和ALKBH5而不是GCLM调节干性。
接下来,作者考虑了GCLM对胶质母细胞瘤生存能力和体内肿瘤生长的贡献。作者用对照shRNA或两个靶向GCLM的非重叠shRNA中的一个转导了两个患者衍生的GSCs。GCLM表达缺失降低了肿瘤细胞活力(图4E)和荷瘤小鼠的存活率(图4F)。
基于ALKBH5调节GCLM m6A修饰的假设,作者进行了甲基化(m6A)RNA免疫沉淀-定量聚合酶链反应(MeRIP qPCR),结果表明ALKBH5敲低增加了GCLM甲基化(图4G)。支持ALKBH5磷酸化在下游靶标调控中的作用,ALKBH5缺失对GCLM m6A修饰的影响通过重新表达ALKBH5WT而不是ALKBH5Y71F完全恢复(图4G),这表明ALKBH5的磷酸化对于GCLM m6A修饰至关重要。然后作者测试了m6A修饰对GCLM表达的作用。通过免疫印迹和免疫荧光检测,在GSCs中靶向ALKBH5可以降低GCLM蛋白水平,通过重新表达ALKBH5WT而不是ALKBH5Y71F,可以完全恢复ALKBH5损失的影响(图4H和4I)。作者探索了m6A修饰如何影响GCLM表达。ALKBH5敲低在两个患者来源的GSCs中降低GCLM mRNA水平(图4J)。由于m6A修饰经常导致m6A修饰mRNA的衰减,而ALKBH5的缺失增加了GCLM上m6A的水平,作者假设GCLM转录物的衰减可能受到ALKBH5的调节。事实上,用shAKBH5转导的GSCs显示出更快的GCLM转录物衰减(图4K)。YTHDF2是一个m6A阅读器,可以诱导mRNA衰变。GSCs的YTHDF2水平高于NSCs。因此,作者使用RBPsuite将YTHDF2结合映射到GCLM转录本上(图4L)。为了测量YTHDF2与GCLM转录本的结合,作者进行了RNA免疫沉淀,然后使用IgG对照或YTHDF2抗体进行qPCR(RIP-qPCR),然后对GCLM进行qPCR。支持ALKBH5在调节YTHDF2结合中的作用,靶向ALKBH5表达的shRNA增强了YTHDF2与GCLM mRNA的结合(图4M)。最后,作者测量了有YTHDF2药理学抑制剂(DC-Y13-27)和没有YTHDF2药理学抑制剂(DC-Y13-27)时,SHAKBH5对GCLM mRNA水平的影响,发现抑制YTHDF2功能可以恢复ALKBH5调节后改变的GCLM水平(图4N)。总的来说,作者的数据表明ALKBH5通过抑制YTHDF2介导的衰退来增加GCLM mRNA水平。
GCLM是GSH合成的限速步骤,因此作者测量了ALKBH5敲除或不敲除GSCs中的GSH水平。ALKBH5敲低降低了总GSH和还原型GSH水平,而ALKBH5WT而不是ALKBH5Y71F的重新表达完全恢复了ALKBH5缺失的影响(图4O)。GCLM作为ALKBH5的下游介质发挥作用,GCLM的表达挽救了体内靶向ALKBH5的作用(图4P和4Q)。ALKBH5共同调节GCLM m6A修饰和蛋白水平,以维持肿瘤生长。
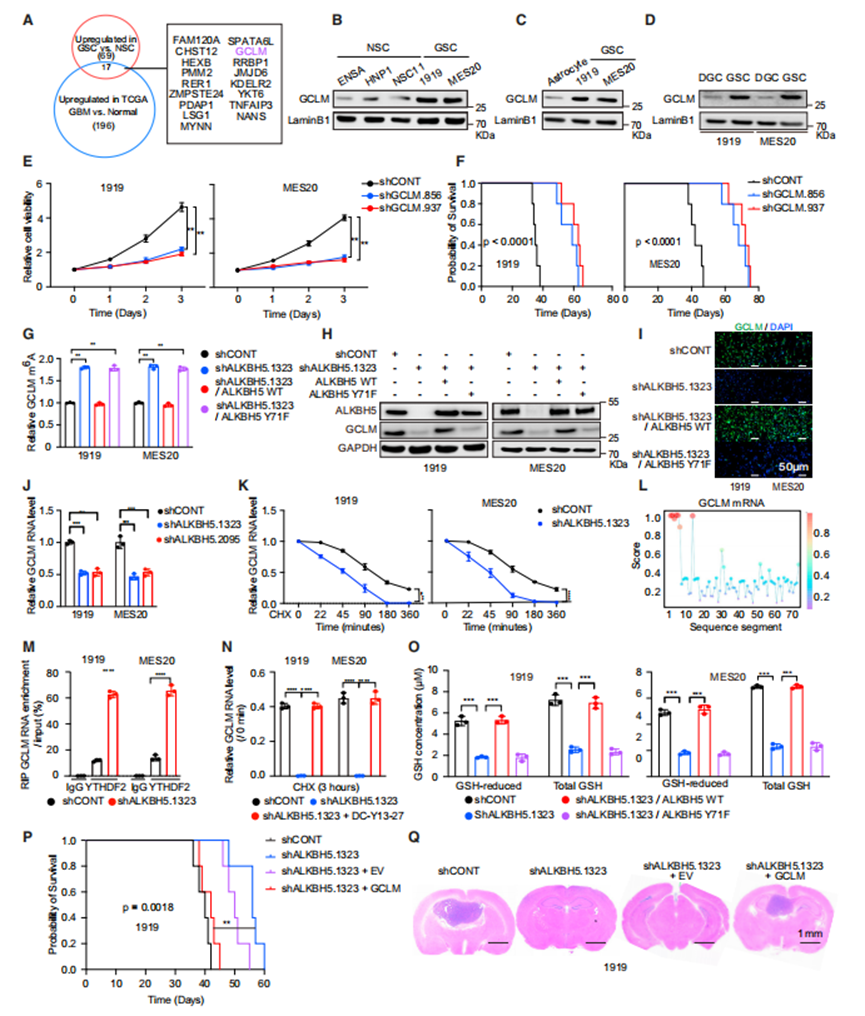 GCLM是ALKBH5在GSCs中的特异性靶点
GCLM是ALKBH5在GSCs中的特异性靶点
5、ALKBH5通过GCLM促进铁死亡存活
铁死亡是一种铁依赖性的细胞死亡形式,由无限制的脂质过氧化和随后的膜损伤引起。活性氧增加和脂质氧化是铁死亡的两个标志。先前的报道支持GSH在铁死亡中的作用,以及GCLM在癌症中铁死亡的调节中的作用。GCLM敲低诱导GSC铁死亡形态和细胞死亡(图5A-5C)。电镜显示,GCLM在GSCs中敲低导致线粒体大小减小,线粒体膜密度增加,线粒体脊丢失(图5D)。靶向ALKBH5表达表型复制了在电子显微镜下观察到的GSC结构改变和诱导细胞死亡(图5E和5F)。为了证明ALKBH5通过调节GSH诱导铁死亡,作者将ALKBH5的表达定位在GSCs中,然后通过提供还原型GSH来测量其拯救作用的能力。ALKBH5敲低降低了细胞活力,增加了SYTOX+细胞群,通过减少GSH完全逆转了这一点(图5G和5H)。靶向ALKBH5表达可诱导ROS和氧化脂质的积累,而通过降低GSH可逆转这一过程(图5I-5L)。因此,EGFR-ALKBH5-GCLM通过GSH调节来保护铁死亡细胞。
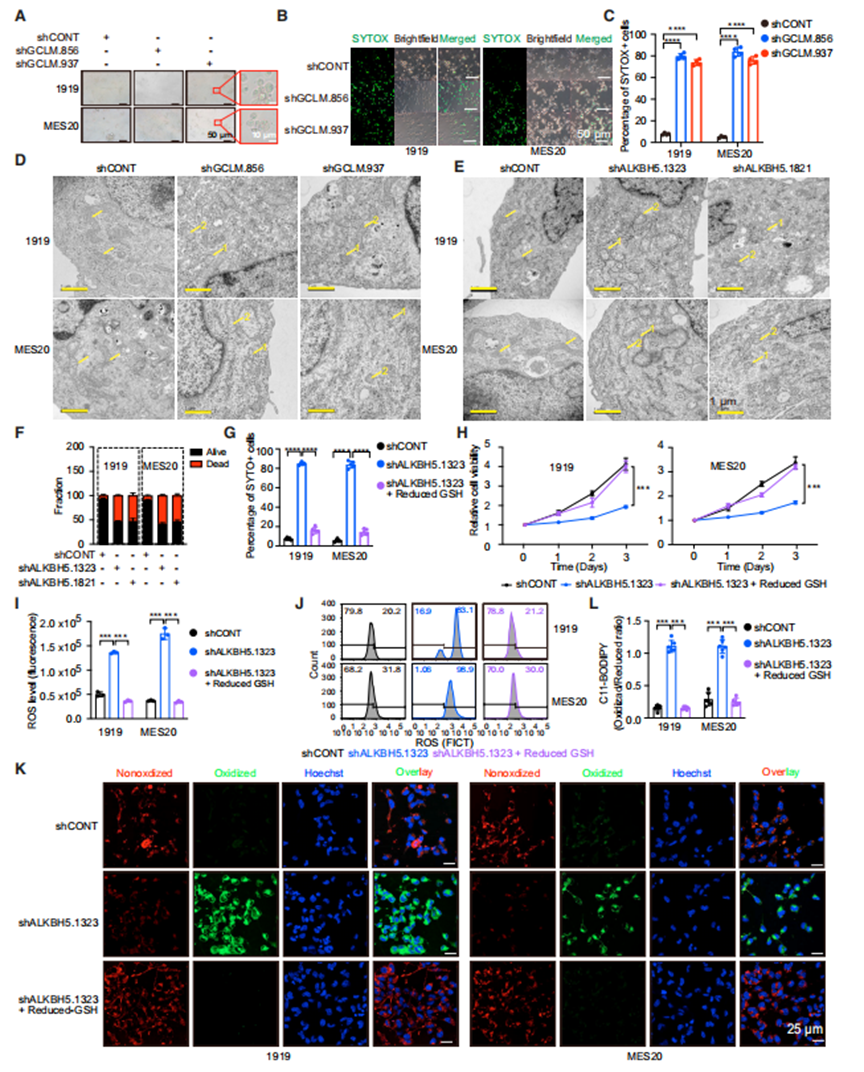 ALKBH5通过GCLM抑制铁死亡
ALKBH5通过GCLM抑制铁死亡
6、ALKBH5的药理学靶向增强了EGFR抑制的抗肿瘤效果
作者选择了美国食品和药物管理局(FDA)批准的EGFR抑制剂(erlotinib)和两种抑制ALKBH5酶活性的化合物(指定为ALKBH5抑制剂1 [ALKBH5i1]和ALKBH5抑制剂2 [ALKBH5i2])(图6A)。使用ALKBH5抑制剂治疗可增加GSCs中的m6A水平,支持ALKBH5抑制剂的靶效应(图6B)。基于EGFR和ALKBH5之间的分子相互作用,作者假设联合这些抑制剂可以提供更大的抗肿瘤效果。ALKBH5抑制剂增强了厄洛替尼对GSCs的疗效(图6C)。
无论是使用ALKBH5i还是厄洛替尼,其疗效与单一药物相似,都能减少肿瘤在体内的生长,而联合治疗能更大程度地减少肿瘤体积(图6D)。每一种药物单独治疗时,肿瘤生长的减少转化为原位荷瘤小鼠的生存期延长,联合治疗时生存率提高(图6E-6G)。为了成为一种可行的脑肿瘤治疗药物,药物需要有一个治疗指数和进入中枢神经系统,因此作者评估了这些药物联合在体内的毒性和分布。用EGFR和ALKBH5抑制剂单独治疗或联合治疗均未引起肝毒性的实验室迹象,通过血清天冬氨酸转氨酶和丙氨酸转氨酶活性水平测量(图6H和6I),也未引起肝、肾或心脏损伤的组织学迹象(图6J)。为了测量药物传递,作者用EGFR或ALKBH5抑制剂治疗患有胶质母细胞瘤的原位异种移植物小鼠,然后作者收集血清、肿瘤和非肿瘤脑来测量药物水平。
接下来,作者基于EGFR和ALKBH5的表达,比较了TCGA和中国胶质瘤基因组图谱(CGGA)中胶质母细胞瘤患者的生存率。肿瘤中EGFR和ALKBH5均高表达的患者预后较低表达的患者差(图6K和6L)。综上所述,ALKBH5与EGFR联合治疗胶质母细胞瘤患者是一个潜在的治疗靶点。
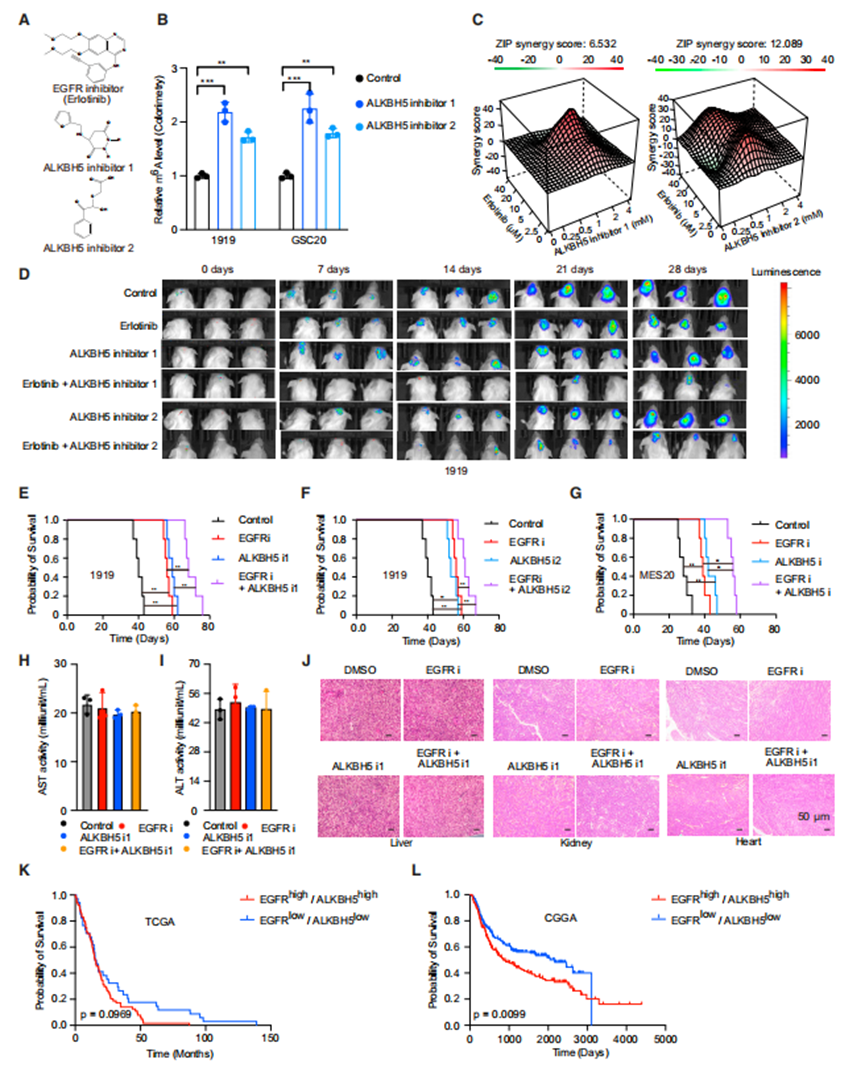 ALKBH5的药理靶向增强了EGFR的抗肿瘤作用
ALKBH5的药理靶向增强了EGFR的抗肿瘤作用
7、ALKBH5的药理学靶向增强了GCLM抑制剂的抗肿瘤功效
诱导铁死亡的药物已被描述为潜在的辅助抗癌治疗。癌细胞具有更高的铁代谢需求,使其比正常细胞更容易发生铁死亡,而GSCs优先运输铁。BSO是一种有效的、特异性的、选择性的不可逆GCLM抑制剂(图7A)。BSO诱导铁死亡,并表现出相对于DGCs对GSCs的优先活性(图7B)。与EGFR和ALKBH5一样,ALKBH5和GCLM似乎是顺序连接的,但大多数分子节点具有多个输入和输出,包括反馈机制。这导致了垂直整合治疗的组合方法,特别是b-raf原癌基因,丝氨酸/苏氨酸激酶(BRAF)和丝裂原活化蛋白激酶(MAPKK, MEK)抑制剂在黑色素瘤中的应用。作者假设联合靶向ALKBH5和GCLM可能会显示出额外的益处。事实上,ALKBH5和GCLM抑制剂在体外联合使用显示出协同抗GSC的功效(图7C和7D)。用生物发光标记转导的胶质母细胞瘤原位异种移植物小鼠接受了载体对照、ALKBH5抑制剂、GCLM抑制剂或联合治疗。单独使用ALKBH5i或GCLM抑制剂治疗可减少体内肿瘤体积,并具有体内联合治疗的额外益处(图7E)。通过ALKBH5和GCLM抑制剂的联合治疗获益,减少肿瘤体积与延长生存期有关(图7F-7H)。
最后,作者考虑了ALKBH5-GCLM、EGFR-GCLM和EGFR-ALKBH5-GCLM的预后意义。为了在没有细胞培养的情况下证实EGFR和GCLM之间的联系,作者询问了胶质母细胞瘤患者标本的空间转录组学和代谢组学数据。EGFR和GCLM mRNA在单细胞水平上相关(图7I),EGFR mRNA水平在空间上与GSH代谢相关(图7J和7K),尽管不是完全相关。综上所述,靶向表观转录组调控和铁死亡联合应用代表了胶质母细胞瘤的潜在治疗模式。
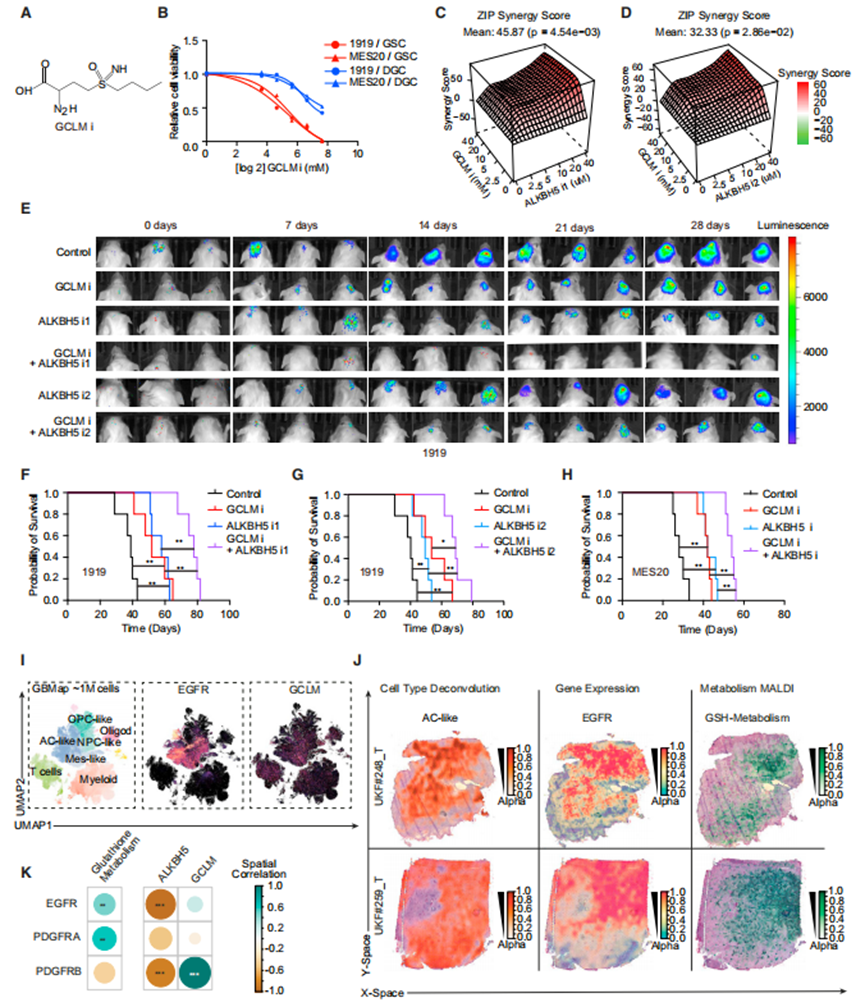 以GCLM为靶点,诱导铁死亡,对GSCs产生抗肿瘤作用
以GCLM为靶点,诱导铁死亡,对GSCs产生抗肿瘤作用
结论:
胶质母细胞瘤在微环境因素和治疗下调节RTKs的下游效应物,这表明同时靶向这些信号通路的多种成分对于避免耐药至关重要。作者的研究结果表明,联合靶向EGFR和ALKBH5是有希望的,厄洛替尼和ALKBH5抑制剂在体外和体内都有联合益处。对铁死亡的敏感性取决于参与ROS、铁、脂质和能量代谢的基因和途径。尽管GSC对传统的铁死亡抑制剂具有耐药性,但GCLM抑制剂选择性地抑制GSC的生长和肿瘤形成。GCLM和ALKBH5抑制剂对GSC生长有协同作用。这些结果表明,靶向该通路中的多个节点可能是有益的,可能表明存在正反馈回路或多重相互作用,需要中断以获得最佳治疗效果。总的来说,这些药物组合值得进一步研究。
实验方法:
GSC提取;细胞培养;质粒分离和定点诱变;逆转录病毒包装与感染;体内肿瘤发生;患者数据库和生物信息学;细胞活性;细胞分级分离;免疫印迹;免疫荧光分析;免疫共沉淀;mRNA纯化;m6A斑点杂交;m6A定量分析;m6A RNA修饰的LC-MS定量分析;LC-MS/MS分析药物浓度;β-半乳糖苷酶细胞染色;ALT活性检测;AST活性检测;MeRIP-qPCR;活性氧检测。
参考文献:
Lv D, Zhong C, Dixit D, Yang K, Wu Q, Godugu B, Prager BC, Zhao G, Wang X, Xie Q, Bao S, He C, Heiland DH, Rosenfeld MG, Rich JN. EGFR promotes ALKBH5 nuclear retention to attenuate N6-methyladenosine and protect against ferroptosis in glioblastoma. Mol Cell. 2023 Nov 8:S1097-2765(23)00862-6. doi: 10.1016/j.molcel.2023.10.025. Epub ahead of print. PMID: 37979586.