血管生成
血管生成是指在原有血管的基础上形成新的血管,它是一个生物过程,可确保维持充足的血流量,为体内细胞提供充足的营养和氧气。大量可溶性生长因子和抑制剂、细胞因子、蛋白酶以及细胞外基质蛋白和粘附分子严格调控着血管生成的多因素过程。人们对血管内皮生长因子(VEGFs)、成纤维细胞生长因子(FGFs)和血管生成素等关键血管生成分子的特性和相互作用进行了深入研究,以了解它们对血管生成的分子影响。自血管生成生长因子被发现以来,许多研究都集中在它们的生物作用上,以及根据病理情况将它们作为血管生成或抗血管生成策略的潜在治疗目标。人们普遍认为,这些因子在血管生成中发挥着不可或缺的作用。
1. 外泌体circCOL1A1通过招募EIF4A3蛋白并激活结直肠癌中的 Smad2/3通路促进血管生成

研究背景:结直肠癌(CRC)是全球第三大高发癌症,发病率和死亡率都很高。我们之前的报告表明,circCOL1A1(hsa_circ_0044556)在CRC中起着癌基因的作用,基因本体(Gene Ontology,GO)分析也揭示了 circCOL1A1与血管生成之间的密切联系。然而,circCOL1A1或外泌体circCOL1A1在CRC血管生成中的作用机制仍不明确。
研究方法:采qRT-PCR、免疫组织化学或Western blot检测circCOL1A1、EIF4A3、Smad通路和血管生成标志物的表达。通过CCK-8检测法监测 HUVECs的细胞增殖。伤口愈合和血管形成试验分别检测了HUVEC的迁移能力和血管生成能力。生物信息学分析、RNA免疫沉淀(RIP)、RNA拉取和FISH检测被用来检测circCOL1A1、EIF4A3和Smad2/3 mRNA之间的相互作用。体外实验结果在异种移植模型中得到了验证。
研究结果:来源于CRC细胞的外泌体circCOL1A1通过招募EIF4A3促进HUVECs的血管生成。EIF4A3在CRC组织中升高,它通过直接结合和稳定Smad2/3 mRNA刺激HUVECs的血管生成。此外,外泌体 circCOL1A1在体外通过诱导Smad2/3信号通路促进血管生成,在体内也加速了肿瘤生长和血管生成。
结论:源自CRC细胞的外泌体circCOL1A1通过招募EIF4A3和激活Smad2/3信号通路促进血管生成。
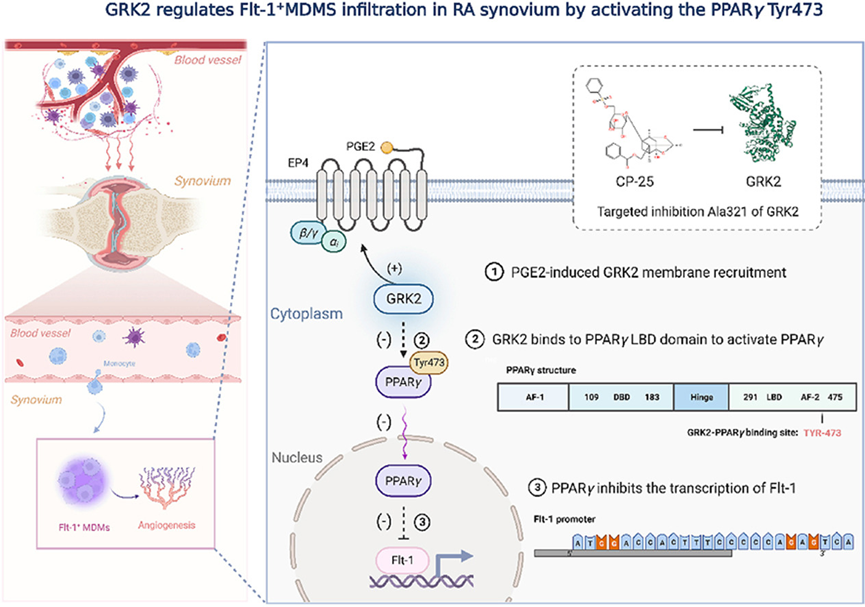
2. GRK2抑制Flt-1+巨噬细胞浸润及其在类风湿性关节炎中的促血管生成特性

类风湿性关节炎(RA)是一种病因复杂的自身免疫性疾病。单核细胞衍生巨噬细胞(MDMs)的浸润与类风湿关节炎的严重程度有关。我们曾报道过,删除G蛋白偶联受体激酶2(GRK2)可通过恢复G蛋白偶联受体信号转导,将巨噬细胞重编程为抗炎表型。然而,随着更多与GRK2相互作用的蛋白被发现,GRK2在RA中的相互作用机制还未得到充分研究。因此,在胶原蛋白诱导的关节炎小鼠模型中,我们使用 GRK2f/fLyz2-Cre+/− 小鼠进行了遗传性GRK2缺失。GRK2f/fLyz2-Cre+/− 小鼠的滑膜炎症和M1极化得到了改善。RNA-seq和双荧光素酶报告实验证实过氧化物酶体增殖激活受体γ(PPARγ)是一种新的与GRK2相互作用的蛋白。我们进一步证实,GRK2-PPARγ信号传导抑制了促进巨噬细胞迁移以诱导血管生成的fms相关酪氨酸激酶1(Flt-1)。从机制上讲,CIA MDMs中过量的GRK2膜募集减少了PPARγ配体结合域的活化,增强了Flt-1的转录。此外,用GRK2活性抑制剂治疗小鼠可显著减轻CIA病理变化、Flt-1+巨噬细胞诱导的滑膜炎症和血管生成。总之,我们预计这将有助于阐明以前未认识到的GRK2特异性细胞内信号转导的细节。靶向GRK2活性是抑制MDMs浸润的可行策略,为控制RA 的关节炎症和血管生成提供了一种独特的方法。
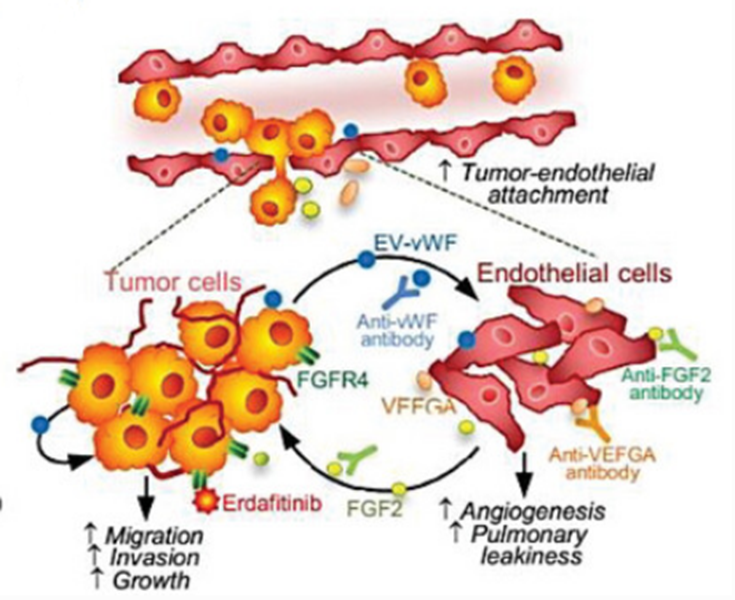
3. 小的细胞外囊泡衍生的vWF诱导肿瘤和内皮细胞之间的正反馈回路,以促进肝细胞癌的血管生成和转移

肝细胞癌(HCC)是一种高血管性恶性肿瘤,其生长和扩散主要受肿瘤衍生的小细胞外囊泡(sEVs)的调控。通过对对照组和HCC患者的循环sEVs进行蛋白质组学分析,发现 von Willibrand 因子(vWF)会随着HCC分期而逐渐上调。与正常人相比,更多的HCC-sEV样本和转移性HCC细胞系中发现了sEV-vWF水平升高。晚期HCC患者循环中的sEV会显著促进血管生成、肿瘤内皮粘附、肺血管渗漏和转移,而抗vWF抗体会明显降低这些作用。血管内皮生长因子A(VEGF-A)和成纤维细胞生长因子2(FGF2)水平的升高可调节内皮细胞。从机制上讲,分泌的FGF2通过 FGFR4/ERK1 信号通路在HCC中引起正反馈反应。在患者异种移植小鼠模型中,联合应用抗vWF抗体或FGFR抑制剂可显著改善索拉非尼的治疗效果。这项研究揭示了肿瘤衍生的sEVs和内皮血管生成因子在HCC和内皮细胞之间的相互刺激,促进了血管生成和转移。它还为新的治疗策略提供了启示。
4. 抑制CXXC5的胞质功能通过增强血管生成和皮肤修复来加速糖尿病伤口愈合

糖尿病伤口愈合,包括糖尿病足溃疡(DFU),是糖尿病的一种严重并发症。考虑到糖尿病足溃疡发生发展的复杂性,确定一种介导多种病因的因子对于治疗非常重要。本研究发现,Wnt/β-catenin通路的负调控因子CXXC型锌指蛋白5(CXXC5)在DFU患者和糖尿病诱导的模型小鼠伤口组织中过度表达,抑制了Wnt/β-catenin通路及其参与伤口愈合和血管生成的靶基因。KY19334是一种通过抑制CXXC5-Dvl 相互作用来激活Wnt/β-catenin通路的小分子,它能加速糖尿病小鼠的伤口愈合。通过恢复被抑制的Wnt/β-catenin信号转导,进而诱导其靶基因,糖尿病小鼠的伤口愈合能力得以增强。此外,KY19334还能诱导后肢缺血模型小鼠的血管生成。总之,这些研究结果表明,通过抑制细胞质CXXC5 的功能来恢复激活Wnt/β-catenin信号可以成为治疗DFU的一种治疗方法。
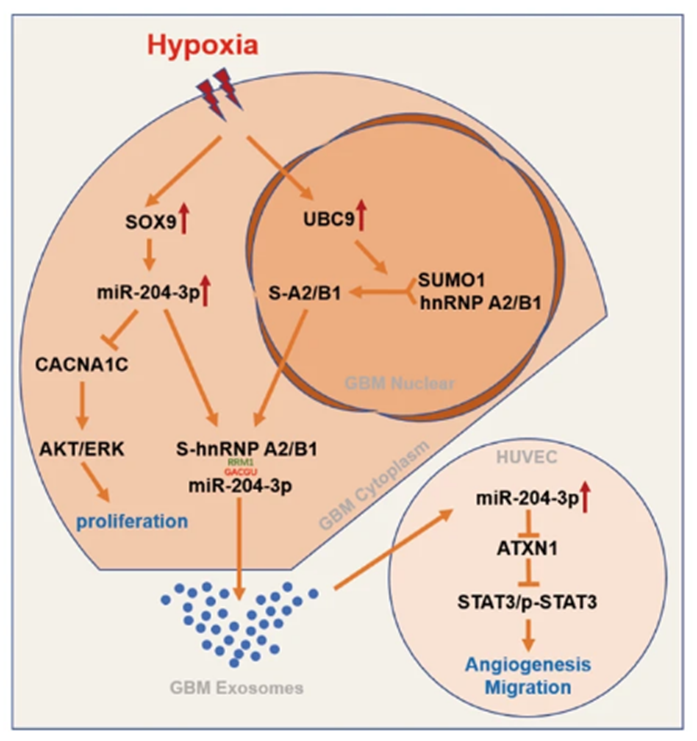
5. 胶质母细胞瘤上调hnRNP A2/B1的SUMO化以消除肿瘤抑制因子 miR-204-3p,从而加速缺氧下的血管生成

胶质瘤是成人中枢神经系统最常见的恶性肿瘤。肿瘤微环境(TME)与胶质瘤患者的不良预后有关。胶质瘤细胞可将miRNA分拣到外泌体中,从而改变TME。缺氧在这一分选过程中发挥了重要作用,但其机制尚不清楚。我们的研究旨在发现分选到胶质瘤外泌体中的miRNA,并揭示其分选过程。对胶质瘤患者脑脊液(CSF)和组织的测序分析表明,miR-204-3p倾向于被分选到外泌体中。miR-204-3p通过
CACNA1C/MAPK途径抑制胶质瘤的增殖。缺氧在miR-204-3p的外泌体分选中起着重要作用。缺氧可通过上调翻译因子SOX9来上调 miR-204-3p。缺氧通过上调hnRNP A2/B1的SUMO化来消除miR-204-3p,从而促进hnRNP A2/B1转移到细胞质。外泌体miR-204-3p通过 ATXN1/STAT3通路促进血管内皮细胞的管形成。SUMO化抑制剂TAK-981可抑制miR-204-3p的外泌体分选过程,从而抑制肿瘤生长和血管生成。本研究发现,胶质瘤细胞在缺氧条件下可通过上调SUMO化消除抑制因子miR-204-3p,从而加速血管生成。SUMO化抑制剂TAK-981 可能是治疗胶质瘤的潜在药物。这项研究发现,胶质瘤细胞在缺氧条件下可通过上调SUMO化消除抑制因子miR-204-3p,从而加速血管生成。SUMO化抑制剂TAK-981可能是治疗胶质瘤的潜在药物。