






肿瘤内皮细胞自噬是黑色素瘤的关键血管免疫检查点
使用免疫检查点阻断剂(ICBs)的免疫治疗在多种实体瘤(包括黑色素瘤)的治疗中显著提高了CD8 T细胞介导的抗肿瘤免疫。然而仍有相当数量的黑色素瘤患者对免疫治疗无效。黑色素瘤是一种典型的免疫原性肿瘤,可以通过多种癌细胞内在和外在机制来维持深刻的免疫抑制性肿瘤微环境(TME)。阐明促进免疫抑制TME的分子机制对于克服黑色素瘤免疫治疗的耐药性具有重要的临床意义。与大多数处于静止状态的健康组织内皮细胞(ECs)相比,TECs不断暴露于导致功能和结构异常的血管生成线索。晚期肿瘤中异常血管生成导致的血管重塑通过增强TECs的免疫抑制特性来对T细胞浸润产生屏障。阐明能够对抗TEC免疫抑制特性的关键机制,或阐明其炎症状态,对于充分释放抗肿瘤免疫应答至关重要。自噬是肿瘤和其他炎症状态中免疫和炎症微环境的重要调节因子。在宿主组织中自噬的丧失会诱导系统性和组织特异性的代谢重编程,从而有利于抗肿瘤免疫。然而自噬如何调节与免疫细胞交界的血管的免疫调节功能尚不清楚。与TEC相关的自噬在何种程度上影响炎症、免疫监视和对ICBs的反应仍然未知。该研究发表在《EMBO Molecular Medicine》,IF:11.1。
技术路线:

主要研究结果:
1. 内皮细胞自噬缺失可改善黑色素瘤的免疫监视
为了揭示内皮细胞自噬在调节黑色素瘤免疫监视中的作用,研究者将可诱导的PDGFb-creERT2Rosa26tdTomato/tdTomato细胞系与ATG5fl/fl小鼠杂交,在他莫昔芬给药后从血液内皮细胞中删除必需的自噬基因Atg5(即ATG5BECKO)。研究者在WT和Atg5BECKO小鼠中比较了皮下移植的小鼠B16-F10黑色素瘤和免疫原性较高的Hcmel12黑色素瘤的生长情况,Hcmel12是在Hgf-Cdk4R24C小鼠中连续移植UV诱导的原发性黑色素瘤。在两个同系模型中,在血液内皮细胞中敲除ATG5使肿瘤负荷达到相似的降幅(图1A-D)。
由于典型自噬蛋白ATG5/ATG12是调节发育中的自噬体的多聚体泛素样结合复合物的一部分,研究者推断ATG12缺失应该是TEC中ATG5缺失导致的功能性上位性改变。ATG9是一种被ULK1磷酸化的跨膜蛋白,并被招募到PI3KC3自噬起始机制复合体,该复合体控制自噬体形成的第一步。此外,虽然Atg5和Atg12都参与了与LC3相关的吞噬作用,但ULK1复合体的成分似乎并未参与其中。在比较接种B16-F10或免疫原性YUMMER 1.7黑色素瘤细胞的野生型和Atg12ECKO或Atg9ECKO小鼠时,研究者观察到肿瘤负荷的类似表型差异(图1E-H)。
在B16-F10和YUMMER 1.7肿瘤中,TEC-自噬缺失均增加了CD31+ TEC周围CD3+ T淋巴细胞的存在,尤其是在血管化程度较高的肿瘤边缘(图1I,J),这提示无论使用的黑色素瘤模型的免疫原性如何,TEC-自噬的缺失均增加了吸引和募集T细胞的能力。
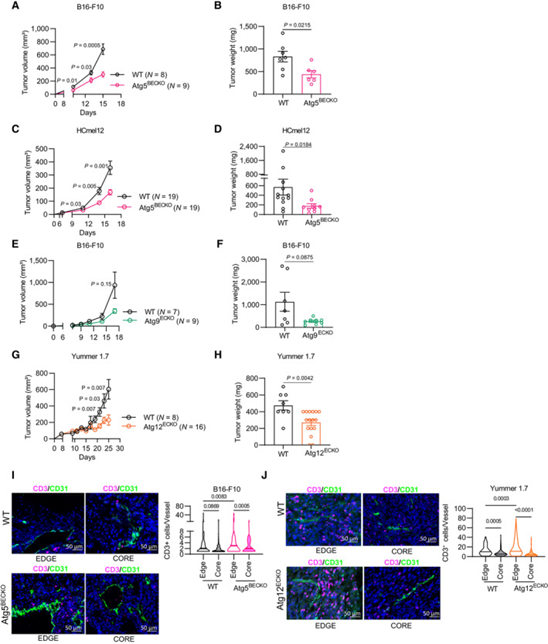
图1 内皮细胞自噬的遗传缺失会降低黑色素瘤的生长
然后,研究者通过FACs分析来自B16-F10 WT和Atg5BECKO小鼠的TIL中CD8+和CD4+ T细胞的存在。与WT小鼠的黑色素瘤相比,Atg5BECKO小鼠的黑色素瘤显示出CD8+ T和CD4+ T细胞数量增加(图2A,B),并且含有关键效应分子颗粒酶B(GrzB)表达较高的CD8+ T细胞(图2A)。携带黑色素瘤的Atg5BECKO小鼠也表现出免疫抑制性T调节性细胞与CD8+T细胞的比例下降,这是TME免疫抑制状态减弱的一个指标(图2C)。
为了进一步了解活化的TIL群体,研究者分析了前体耗竭的CD8+ T细胞,其标志是TCF1和PD1的联合表达,以及缺乏TIM3表达,以及其后代的终末耗竭效应因子PD1+ CD8+ T细胞,定义为GrzB增加和TIM3水平高,但缺乏TCF1表达。在WT和Atg5BECKO小鼠中,两个种群保持相似(图2D)。然而,与WT小鼠相比,来自Atg5BECKO的最终耗竭效应性CD8+ T细胞产生的GrzB量较高(图2E)。
然后,研究者询问了在ATG5BECKO小鼠中,CD8+ T细胞增加和肿瘤生长减少之间是否存在因果关系。研究者通过注射αCD8+抗体去除CD8+ T细胞,并在WT和Atg5BECKO小鼠中检测了肿瘤生长(图2F)。去除CD8+ T细胞未显著影响WT小鼠中的B16-F10生长,但逆转了Atg5BECKO小鼠中对肿瘤负荷和生存期的有益作用(图2G,H),因此在功能上提示CD8+T细胞参与延迟的肿瘤生长表型。
总之,这些数据表明,TECs中典型自噬基因的缺失通过增加表达GrzB的效应性CD8+ T细胞的频率来抑制黑色素瘤的生长和具有免疫抑制性的TME。
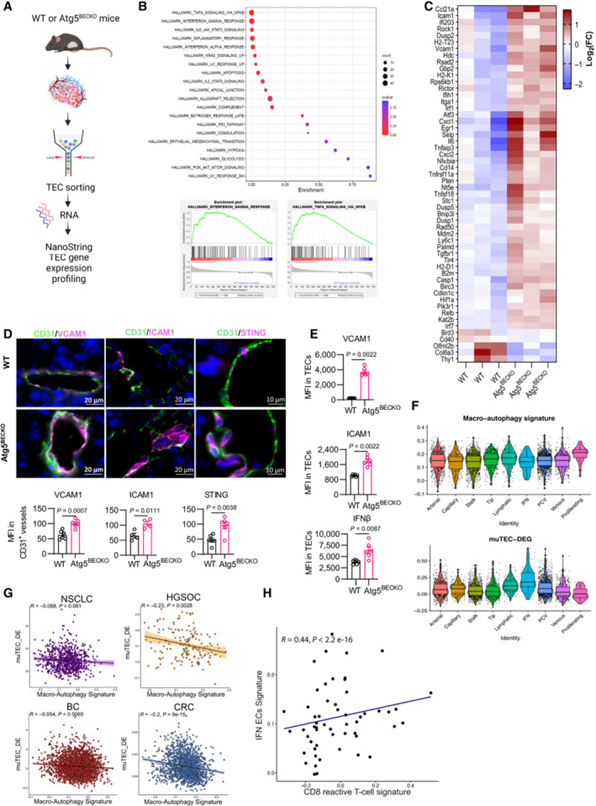
图2 肿瘤内皮细胞中Atg5的缺失促进CD8+ T细胞介导的抗肿瘤免疫反应
2. 自噬促进TEC的免疫耐受状态
接下来,研究者探讨了自噬如何调节淋巴细胞内流和活性,重点关注调节淋巴细胞的趋化因子、细胞因子和黏附分子。自噬不仅通过蛋白质降解发挥直接作用,还调节细胞的转录和表观遗传程序,研究者首先对来自B16-F10肿瘤的FAC分选的Atg5精通和Atg5缺陷的CD45-Ter119-CD31+TEC进行了转录谱分析,使用Nanostring技术对涉及癌症免疫调节的770个基因进行了分析(图3A)。在研究者的数据集中,最主要的特征与炎症反应相关(图3B)。此外,对Log2FC≥0.7且P<0.05的差异表达(DE)基因所做的分析表明,在Atg5BECKO TEC中,编码具有免疫细胞相互作用活性的表面炎症蛋白和细胞因子/趋化因子的基因显著表达,而这些是炎症TEC表型的特征(图3C)。与此同时,与NF-κB通路相关的因子和各种EC黏附分子的基因表达在Atg5BECKO小鼠TECs中显著上调。后者包括VCAM1,ICAM1,SELE和SELP,它们在结合T细胞表面配体和增加T细胞受体信号传导中具有确定的作用,以及在内皮细胞中具有趋化和黏附功能的细胞因子和趋化因子,如IL6、CX3CL1/fractalkine、CXCL2、CXCL1和非经典NF-κB通路的介质RELB(图3C)。另外一组用于抗病毒/1型干扰素(IFN)应答的DE基因包括IFIH1、IRF1、IRF7、TLR4以及抗原呈递机制H2-K1、H2-T23和H2-D1的元件,这提示Atg5BECKO小鼠的TECs获得了改善的免疫调节功能,包括维持T细胞功能的能力(图3C)。基因集富集分析证实,与来自WT小鼠的TECs相比,来自Atg5BECKO小鼠的TECs在参与炎症反应和IFN信号通路的基因集中表现出稳健的富集(图3B)。因此,ATG5敲除引起TEC中控制淋巴细胞黏附和功能的趋化因子和分子的表达。
然后,研究者在选定的免疫调节标志物的蛋白质水平上验证了转录组结果。与WT相比,来自Atg5BECKO小鼠的TECs显示VCAM1、ICAM1和STING(抗病毒/1型干扰素应答的主要调节因子)水平显著增加(图3D)。与此同时,Atg5BECKO小鼠TECs的VCAM1、ICAM1、MHCⅰ类和MHCⅱ类的表面表达以及IFNβ(STING通路的下游效应因子)的胞内表达均升高(图3E)。因此,在各种黑色素瘤模型中,自噬阻断可促进TEC炎症。
接下来,研究者试图通过分析公开获得的单细胞RNA-seq癌症图谱中的ECs表型特征,将研究者的小鼠mRNA谱与人类TECs(huTECs)进行更广泛的背景分析。为了增加罕见huTEC亚型的覆盖率,研究者汇总了来自不同活检部位(原发肿瘤、转移和邻近非肿瘤组织样本)的非小细胞肺癌、乳腺癌、高级别浆液性卵巢癌和结直肠癌的EC scRNAseq数据。从总共7573个高质量TEC开始,研究者对这些肿瘤进行了聚类分析。研究者验证了来自纳米串分析的“炎症”基因标签包含在小鼠和人类之间保守的基因,并使用该内部生成的标签(称为muTEC-DE)和反应组-巨自噬标签进行进一步分析。值得注意的是,以muTEC-DE标签表达最高为标志的IFN huTEC亚群在自噬标签中的富集程度最低(图3F),这提示,特别是免疫调节功能更明显的亚型同时显示出自噬基因的低表达水平。
研究者进一步在各种癌症类型的原发肿瘤huTEC内检验了muTEC-DE和自噬标签之间的关联。值得注意的是,除了非小细胞肺癌之外,研究者在分析的所有其他癌症类型中观察到这些标签之间存在显著的负相关,尽管在乳腺癌中这种负相关程度有所降低(图3G)。此外,huTECs中muTEC-DE标签的富集与T细胞内测定的CD8+反应性T细胞标签的表达呈正相关(图3H)。
综上所述,这些观察结果提示,无论物种或肿瘤类型,自噬能力低的TECs均具有增强的免疫支持功能。
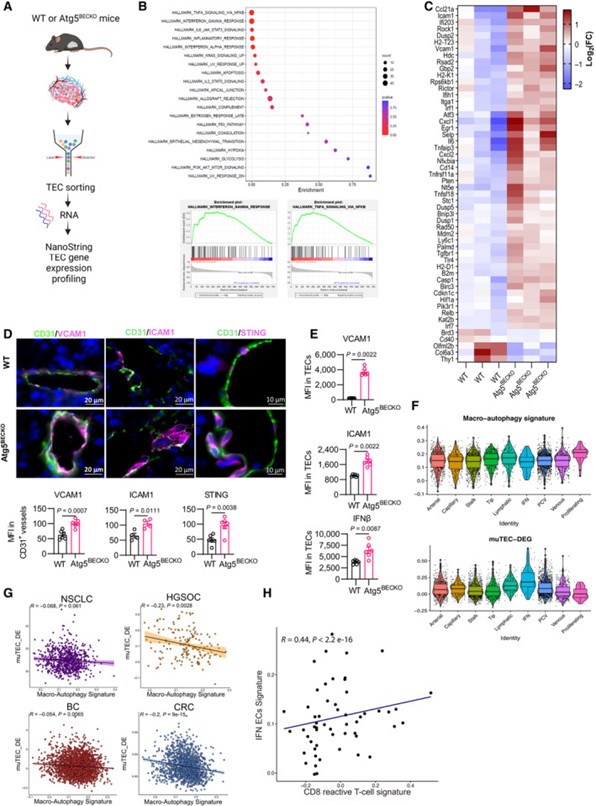
图3 自噬抑制肿瘤内皮细胞的炎性表型
3. 自噬可减弱内皮细胞中NF-κB和cGAS的炎症轴
接下来,研究者的目标是确定自噬抑制支持EC炎症表型的潜在机制。研究者评估了培养的人脐静脉内皮细胞是否可以重现在小鼠中观察到的自噬遗传缺失的影响。与对照内皮(Ctr)相比,Atg5KO内皮中一组基本相似的炎症基因(例如SELE、VCAM1和ICAM1)和趋化因子(例如CX3CL1、CXCL10)的表达增强,这是在对从Atg5BECKO小鼠分离出的muTEC进行的纳米串分析中观察到的结果(图4A-E)。炎症基因表达谱程度的差异(图4A-E)可能可以通过这些细胞中ULK1/2的急性抑制而非ATG5的慢性缺失来解释。Atg5KO细胞中VCAM1的表面表达增加通过FACs进一步验证(图4F)。
考虑到体内和体外表型的相似性,研究者使用培养的自噬缺失的内皮细胞来研究加剧炎症信号传导的分子通路。研究者专注于STING和NF-κB信号通路,因为已知STING激活可通过协同调节I型IFN和NF-κB激活来促进抗肿瘤免疫,而在携带黑色素瘤的Atg5BECKO小鼠的muTEC中增强(图3C,D)。此外,虽然已知自噬可调节STING和NF-κB通路,但在内皮细胞中,去除STING可改善T细胞在TNF诱导的炎症反应中的募集。
在经典的STING激活途径中,cGAS与胞质内的dsDNA结合并催化cGAMP的合成。cGAMP与ER相关STING的结合触发其寡聚化,并从内质网(ER)出口/释放到ER-高尔基中间室,其中寡聚化的STING介导了TBK1的激活/磷酸化。这进而导致干扰素刺激基因的转录和NF-κB的激活。信号停止是通过将STING从后高尔基体贩运到溶酶体来协调的,在溶酶体中,STING被降解。
研究者首先测量了自噬/溶酶体降解途径的干扰对STING运输和信号传导的影响。在静息条件下,通过ATG5KO阻断自噬(图4G)导致STING的单体和二聚体/寡聚体形式逐渐增加,这是通过在非变性条件下的免疫印迹发现的。同样,在ATG5KO ECs中,STING与LMAN1的共定位增加,这与TBK1磷酸化相关(图4H),提示其激活。与此同时,向ECs中添加cGAS酶活性的产物cGAMP,会轻微但不显著地提高Ctr ECs中的p-TBK1和VCAM1,而对ATG5KO ECs没有额外的影响(图4H)。
研究者进一步探索了自噬可以刺激清除cGAS激活因子的可能性,例如来自线粒体的胞质双链DNA(dsDNA),这是STING信号通路的一个已确定的内源性触发因素。在真正凋亡信号驱动的线粒体外膜通透化的细胞中,自噬可以抑制I型IFN。使用TOMM20对线粒体网络进行染色的超分辨率显微镜检查和抗dsDNA抗体显示,与显示在线粒体网络内包裹的dsDNA的Ctr ec相比,ATG5-KO EC中的dsDNA主要位于线粒体网络附近,部分位于细胞质中(图4I),这表明其通过线粒体源性囊泡流出。虽然与Ctr ECs相比,ECs中的ATG5 KO未引起关键线粒体形态测量参数的明显变化(图4J)。此外,鉴于STING含有一个LC3相互作用区域,因此可以作为自噬降解的靶点,研究者不能排除这一机制导致了在ATG5缺失的内皮细胞中观察到的STING信号。尽管如此,这些结果表明,在内皮细胞中自噬的缺失介导了cGAS-STING和NF-κB的激活,导致了一系列炎症/免疫调节靶基因的表达。同样,内皮细胞中ATG5的消融,伴随着VCAM1的上调,导致了经典NF-κB通路的抑制性IκBα蛋白的磷酸化,并增加了替代NF-κB信号通路的介质积累;前体蛋白p100、其降解产物p52和RelB,提示NF-κB通路的激活(图4K)。
然后,研究者评估了在自噬缺失的内皮细胞中,是否需要cGAS-STING来激活NF-κB。为此,研究者利用CRISPR-Cas9在Ctr和ATG5KO细胞中删除了STING。然而,ATG5和STING的伴随和慢性抑制的效应在不同的人类EC供体中不同,这使得结果不可靠。因此,研究者测试了抑制ULK1/2在STING去除细胞中的作用。与ATG5KO类似,抑制ULK1/总之,这些数据假设STING促进了NF-κb依赖的免疫支持Atg5BECKO表型的加重,但不是必需的。
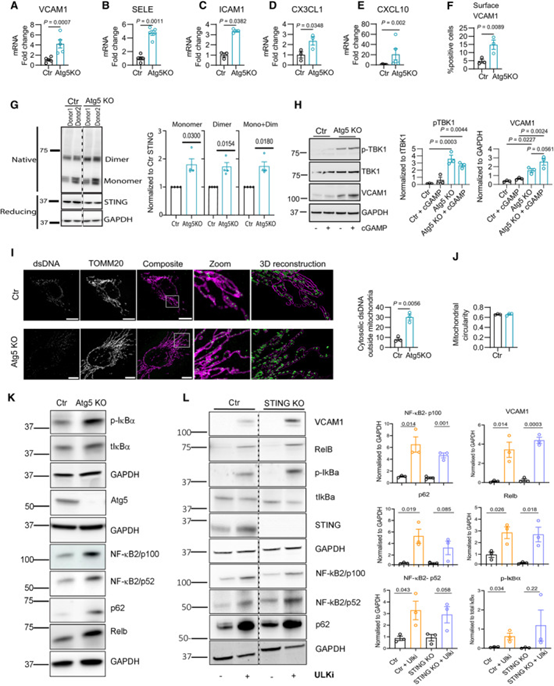
图4 在体外培养的内皮细胞中,自噬通过清除胞质内的双链DNA,通过CGAS-STING-介导的NF-κB通路抑制炎症反应
4. 肿瘤内皮细胞自噬限制黑色素瘤对抗PD1治疗的反应
鉴于阻断自噬具有免疫刺激作用,同时CD8 T细胞浸润和活性增强,因此研究者研究了破坏TEC中的自噬可否通过免疫检查点阻断(ICB)的形式促进免疫治疗(图6A)。虽然抗PD1单药治疗显著降低了WT小鼠中的B16-F10肿瘤生长,但免疫治疗对携带B16-F10的Atg5BECKO小鼠的效果是递增的,但不显著(图6B,C)。这些结果提示,在携带黑色素瘤小鼠的TEC中,阻断自噬有利于TILs的募集和活性(图2D-G),因此并未进一步增强抗PD1 ICB治疗的疗效,但可能进一步维持其疗效。
研究者之前的分析特别表明,treatment-naïve癌症患者的huTEC干扰素亚型在muTEC-DE High和自噬low基因标签的表达之间表现出不同的关联(图3F)。为了进一步阐明这些TEC特异性标签的意义,以及它们与黑色素瘤患者对ICBs的临床应答之间的关联,研究者随后使用来自一项独特的前瞻性纵向研究的scRNAseq数据研究了huTEC的单细胞转录组。在治疗前和第一个治疗周期后收集肿瘤活检组织并进行scRNAseq分析。
数据包括来自22个样本的TEC,N=12有反应(R),N=10无反应(NR)(图6D)。总体而言,研究者使用已建立的无偏倚EC识别方法对1541个EC进行了注释。与无应答者相比,应答者的huTEC在治疗前显著富集了核心炎症基因集(图6E),而自噬基因核心标签的富集较低。有趣的是,无应答者在一个治疗周期后也显示出muTEC-DE标签的富集(图6E)。这可能提示TEC炎症表型与TME的初始变化相关,这一变化是由抗PD1的首次应答驱动。然而,在同一组患者中,较高的自噬评分无变化(图6E),这提示只有muTEC High / autophagy Low状态与临床获益相关。
然后,研究者研究了应答者和无应答者之间的muTEC-DE标签是否仅在huTECs亚型中存在差异。为此,研究者将所有huTECs聚类,并获得了11种亚型,这些亚型反映了在原发泛癌数据中观察到的huTECs异质性。与无应答的黑色素瘤患者相比,对抗PD1有应答的黑色素瘤患者在干扰素、Tip/Stalk和静脉EC簇中有显著富集的muTEC-DE特征,这一趋势在干扰素亚型的ON治疗时间点稳定维持。值得注意的是,应答者中的这些相同簇在ON治疗时间点显示自噬标记的表达显著降低(图6F)。此外,利用这些scRNAseq数据集,研究者发现所有TECs中的muTEC-DE标签表达水平与CD8+T细胞的瘤内浸润呈正相关(图6G)。
总之,这些数据提示,muTEC-DE High/自噬低表型在临床上与抗PD1治疗的良好应答相关。
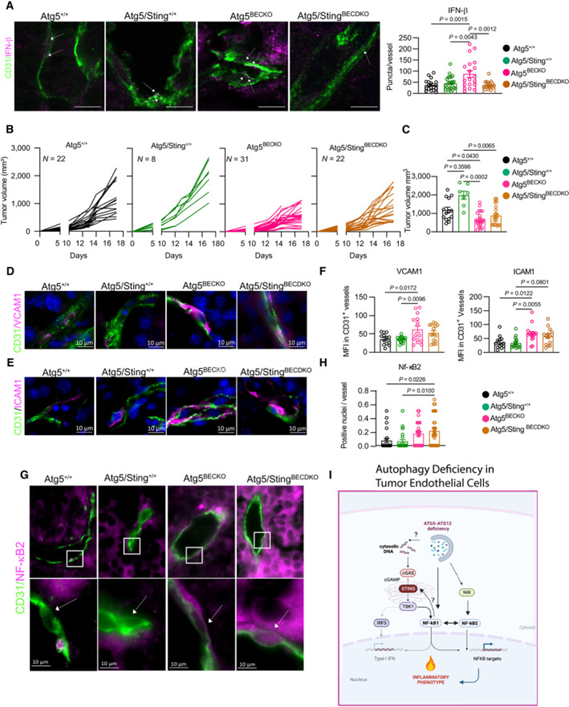
图5 TECs中Sting和Atg5的双重基因缺失促进了替代NF-κB通路的激活
5. 炎症血管和CD8+ T细胞的空间接近与抗PD1治疗的反应相关
然后,研究者试图描述炎症huTEC与黑色素瘤样本中CD8+T细胞和VCAM1或VCAM1/STING蛋白表达增强的空间关系。研究者采用多重迭代标记抗体新沉积技术进行了多重免疫组织化学,研究者使用了接受抗PD1单药治疗的黑色素瘤患者的一组真实生活活检。黑色素瘤活检组包括6例有反应者和6例无反应者。研究者在活检的三个不同空间区室(即肿瘤区、肿瘤-间质界面和非肿瘤区域)评估了CD31/AQP1双阳性(识别huTECs)、CD8+ PD1-GrzB-T细胞(识别naïve CD8+ T细胞)和CD8+ PD1+GrzB+(识别活化/效应性CD8+ T细胞)。在肿瘤体积和肿瘤-间质界面,无反应和有反应的EC数量没有差异(图7A,B)。然后,研究者通过邻域分析研究了这些细胞类型在这些空间区室中的邻近性。与无应答患者相比,缓解患者的VCAM1+和VCAM1+STING+TECs分别在肿瘤区域和肿瘤-间质界面显著富集(图7C,D)。进一步的邻域分析表明,在CD8+ PD1-GrZB-和CD8+ PD1+GrZB+T细胞附近,缓解患者的VCAM1+STING+TECs数量显著较高(图7E,F)。在肿瘤-间质界面和肿瘤区域,这一观察结果是正确的。尤其是在肿瘤体积内,即使数量较低,VCAM1+血管仍然显著接近CD8+ T细胞的两个亚群,这提示即使在没有STING的情况下,炎症血管表型与浸润的TILs保持更良好的交互作用(图7G,H)。研究者也在MILAN分析中使用了LC3抗体,但由于TECs中LC3染色的细胞内颗粒状和弥散型的分辨率有限,研究者无法可靠地评估其相关的自噬状态。
总之,这些结果表明,炎症血管与浸润的CD8+ T细胞之间的空间接近是黑色素瘤患者对抗PD1治疗产生应答的TME的标志。
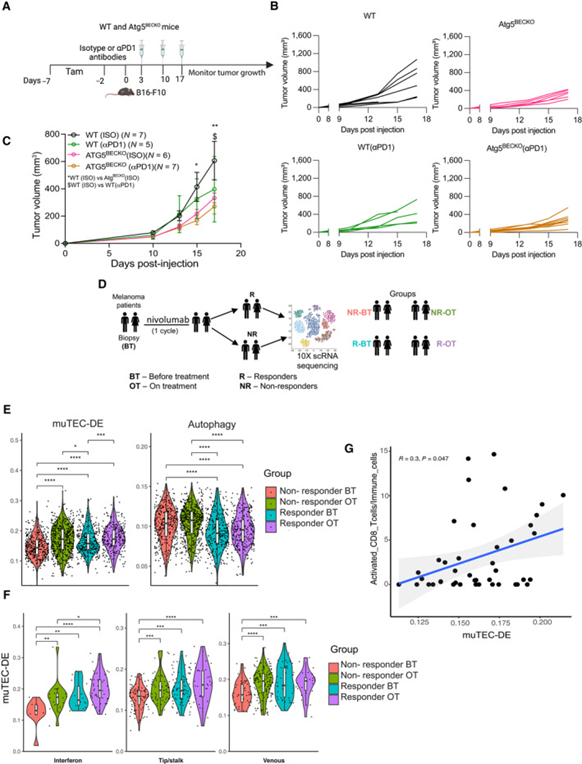
图6 黑色素瘤中TEC‐自噬与抗‐PD1治疗的反应呈负相关
结论:
该研究提高了对自噬作为血管固有的抗炎/免疫抑制机制的作用的认识,限制了黑色素瘤的抗肿瘤免疫。该研究还为利用血管归巢工具确定针对肿瘤血管的自噬抑制剂或NF-κB调节剂的适当联合治疗提供了理论基础。
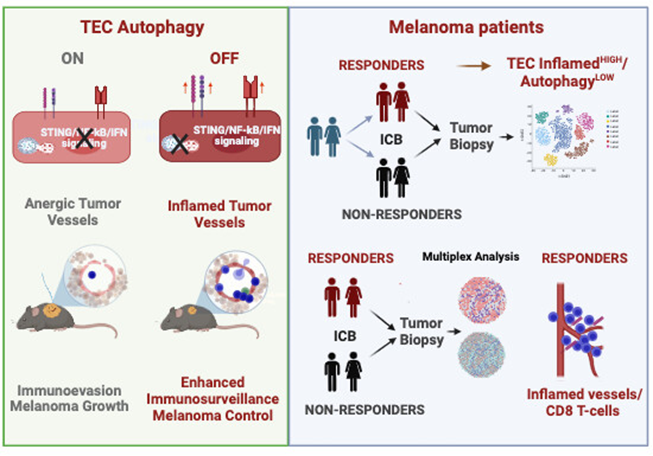
实验方法:
细胞培养,小鼠实验,免疫表型分析,免疫荧光成像,RNA分离,定量RT-PCR,蛋白质印迹,单细胞RNA测序
参考文献:
Verhoeven J, Jacobs KA, Rizzollo F, Lodi F, Hua Y, Poźniak J, et al. Tumor endothelial cell autophagy is a key vascular-immune checkpoint in melanoma. EMBO Mol Med. 2023 Nov 27:e18028. doi: 10.15252/emmm.202318028.