






PRMT3介导的METTL14精氨酸甲基化促进子宫内膜癌的恶性进展和治疗耐受性
蛋白精氨酸甲基转移酶(PRMT)在肿瘤的发生和发展过程中起着至关重要的作用,但其影响子宫内膜癌(EC)治疗敏感性的内在机制仍不清楚,值得进一步研究。本文对癌症基因组图谱数据库和临床蛋白质组肿瘤分析联盟数据库进行了全面分析,发现PRMT3在子宫内膜癌中发挥着重要作用。具体来说,进一步的实验表明,PRMT3抑制会增强EC细胞对铁死亡的敏感性。从机理上讲,PRMT3与甲基转移酶14(METTL14)相互作用,并参与其精氨酸甲基化。此外,PRMT3抑制介导的METTL14过表达会通过m6A-YTHDF2依赖性机制促进甲基化修饰,降低谷胱甘肽过氧化物酶4(GPX4)mRNA的稳定性,增加脂质过氧化水平,加速铁死亡。值得注意的是,PRMT3阻断和抗PD-1联合疗法通过加速细胞衍生的异种移植模型中的铁死亡而显示出更有效的抗肿瘤作用。特异性PRMT3抑制剂SGC707在患者来源的异种移植模型中也发挥了同样的免疫治疗增敏作用。值得注意的是,阻断PRMT3能改善顺铂和放射治疗对肿瘤的抑制作用。总之,这项研究表明,PRMT3是一种很有前景的EC靶点。本文于2023年11月发表于《Advanced Science》,IF=15.1。
技术路线
实验结果
1. PRMT3是EC中一种关键的上调生物标志物
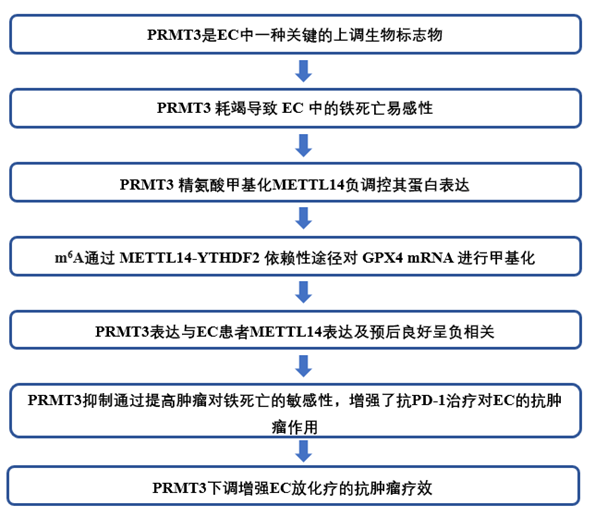
为了确定在EC中起关键作用的PRMTs,作者对来自TCGA数据库和CPTAC数据库的UCEC数据集进行了差异分析,发现在重叠结果中,PRMT1和PRMT3在EC组织中升高(图1A)。qRT-PCR检测证实,在5个临床EC组织样本中,PRMT1和PRMT3的表达均高于匹配的癌旁组织(图1B)。此外,与正常子宫内膜上皮细胞相比,PRMT3在EC细胞中上调。在EC细胞系中,PRMT3在HEC-1A细胞中表达最高,在HEC-1B细胞中表达最低(图1C-E)。
为了进一步确定PRMT3的作用,作者通过慢病毒转染建立了稳定的HEC-1B-Lv-PRMT3和HEC-1A-Lv-shPRMT3细胞。通过Western印迹和qRT-PCR验证了稳定基因转移的有效性(图1F-G)。与对照组相比,PRMT3的上调提高了HEC-1B细胞的存活率,而PRMT3缺陷的HEC-1A细胞的存活率则降低了(图1H)。此外,与对照组相比,过表达PRMT3增加了HEC-1B细胞和HEC-1A细胞的集落形成能力。相比之下,在HEC-1B细胞和HEC-1A细胞中去除PRMT3则观察到相反的趋势(图1I)。此外,过表达PRMT3会显著降低细胞死亡的比例,而敲除PRMT3则会增加这一比例(图1J)。上述发现意味着PRMT3在EC中表达上调,并可能在EC的进展中发挥重要作用。
2. PRMT3耗竭导致EC中的铁死亡易感性
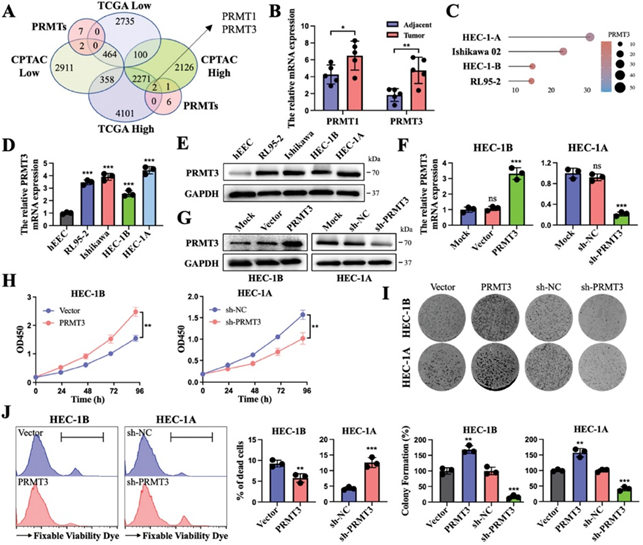
对细胞死亡不敏感是癌症的关键特性之一,它使肿瘤对治疗方法产生部分抗性。用不同浓度的铁死亡诱导剂处理PRMT3下调或对照的EC细胞表明,PRMT3缺失抑制了对弹性蛋白诱导的细胞死亡的抵抗力(图2A)。PRMT3阻断组和Erastin处理组的细胞都表现出一定程度的铁死亡。此外,铁死亡抑制剂liproxstatin-1挽救了这种由 PRMT3敲低促进的Erastin诱导的细胞死亡,但不能被细胞凋亡抑制剂(Z-VAD-FMK)、坏死抑制剂(坏死磺胺)或自噬抑制剂(3-甲基腺嘌呤)所挽救(图2B、C)。这些结果表明,PRMT3的缺失通过增加细胞内的铁死亡而增强了Erastin治疗的抑制作用。
由于GPX4是铁死亡过程的一个显著标志,因此进行了进一步的实验。过表达 PRMT3在蛋白和mRNA水平上都促进了GPX4的表达(图2D)。值得注意的是,GPX4的上调或抑制对PRMT3的表达没有影响。PRMT3作为GPX4的上游因子发挥着调控作用(图2E)。接下来,作者试图研究PRMT3和GPX4对EC中铁死亡敏感性的影响。CCK8测定显示,PRMT3过表达细胞和GPX4过表达细胞的细胞活力增强,而PRMT3缺陷细胞和GPX4缺陷细胞的细胞活力则相反。GPX4的抑制或上调分别部分逆转了PRMT3过表达或敲除诱导的细胞活性变化(图2F)。随后,PRMT3过表达细胞和GPX4过表达细胞中的丙二醛(MDA)和亚铁(Fe2+)水平降低,而PRMT3缺失细胞和GPX4敲除细胞中的丙二醛和亚铁水平升高。PRMT3过表达导致的MDA和Fe2+水平降低或PRMT3缺失导致的MDA和Fe2+水平升高趋势可分别被GPX4抑制或过表达所挽救(图2G,H)。具有代表性的透射电子显微镜图像显示,PRMT3缺失细胞和GPX4缺失细胞的线粒体变小,膜密度增加,这是铁死亡的特征。GPX4的下调或上调分别部分改变了PRMT3的上调或下调所诱导的细胞内铁死亡的程度(图2I)。总之,这些数据表明,沉默PRMT3可以通过抑制EC中GPX4的表达来抑制癌细胞对铁死亡的抵抗力。
3. PRMT3精氨酸甲基化METTL14负调控其蛋白表达
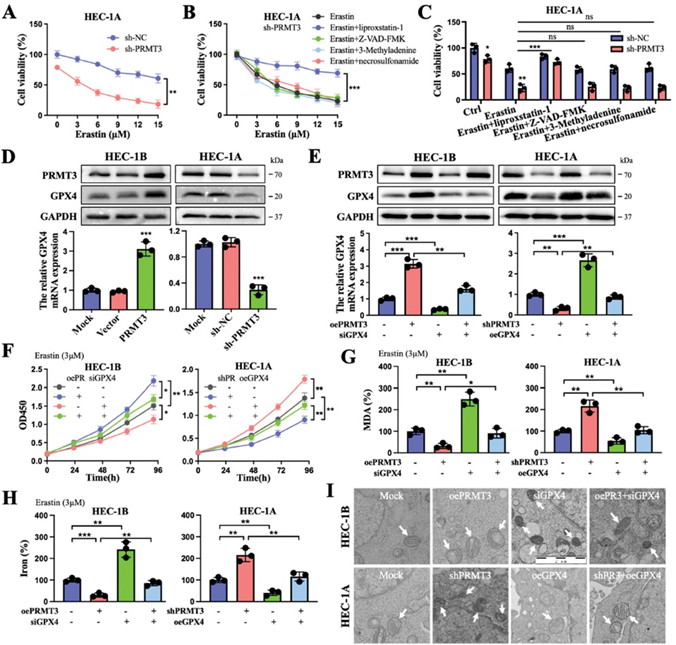
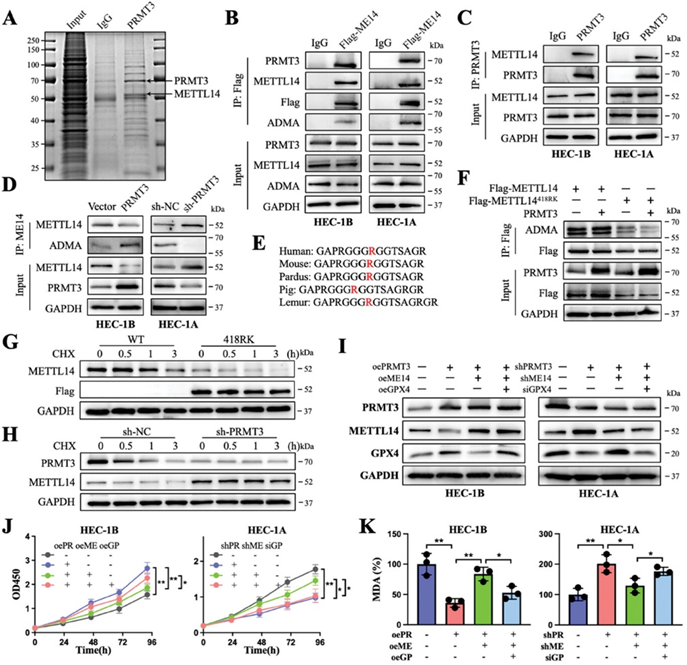
为了阐明PRMT3调节GPX4的机制,作者对PRMT3结合蛋白进行了质谱分析,其中METTL14被认为是可能与PRMT3相互作用的蛋白(图3A)。随后,证实PRMT3与METTL14的结合,并观察到PRMT3催化了METTL14的ADMA(图3B、C)。进一步分析表明,PRMT3能放大METTL14的ADMA信号,而在PRMT3缺失的细胞中该信号被抑制(图3D)。这一精氨酸残基序列在不同物种间的高度保守性表明,这种甲基化可能具有生物学功能(图3E)。此外,作者根据GPS-MSP分析估计了可能的精氨酸残基位置(R418)(http://msp.biocuckoo.org/)。METTL14中R418的赖氨酸突变(418RK)导致METTL14的ADMA甲基化显著消失,表明该位点可能是 PRMT3对METTL14进行精氨酸甲基化的靶点(图3F)。此外,该突变缩短了 METTL14的半衰期,PRMT3缺失阻止了METTL14蛋白的降解(图3G、H)。拯救实验进一步揭示了PRMT3-METTL14-GPX4上下游调控轴之间的蛋白表达和铁死亡之间的调控关系(图3I-K)。总之,PRMT3促进了METTL14的精氨酸甲基化并对其进行表观遗传调控,从而介导了对细胞铁死亡的抑制。
4. m6A通过METTL14-YTHDF2依赖性途径对GPX4 mRNA进行甲基化
METTL14作为甲基转移酶的核心成分,可以参与m6A修饰的动态过程,因此作者进一步研究了METTL14对GPX4的m6A修饰。本节的实验选择了HEC-1A和 Ishikawa细胞,因为它们分别被鉴定为METTL14表达较低和较高的细胞(图4A)。Western印迹和免疫荧光检测显示,METTL14高表达细胞中GPX4表达减少,而 METTL14缺失细胞中GPX4表达增加(图4B)。如结果所示,过表达METTL14显著提高了细胞的全局m6A水平和GPX4 mRNA的m6A富集(图4C、D)。接下来,SRAMP程序(http://www.cuilab.cn/sramp)确定了GPX4 mRNA上潜在的m6A修饰位点。生成包含野生型(WT)或突变型(Mut)GPX4 的荧光素酶报告基因,用于后续检测。在突变型GPX4中,m6A共序中的腺苷核苷酸被胸苷取代(图4E)。含有GPX4-WT的构建体的荧光素酶活性在METTL14过表达时显著降低,而在METTL14缺失时则增强,而GPX4-MUT的荧光素酶活性似乎不受影响(图4F)。这些结果表明 GPX4的表达受METTL14介导的m6A修饰控制。
考虑到m6A甲基化过程涉及m6A阅读蛋白的识别,研究人员调查了YTHDF1/2/3作为经典的m6A阅读蛋白家族对GPX4 mRNA稳定性的影响。研究发现,YTHDF2缺失后,GPX4的表达升高;然而,在YTHDF1/3缺失的细胞中,GPX4没有发生变化(图4G,H)。YTHDF2缺乏消除了METTL14过表达对GPX4的抑制作用(图4I)。RIP-qPCR证实了YTHDF2和GPX4 mRNA之间的相互作用(图4J)。此外,与使用放线菌素D处理后的对照细胞相比,METTL14基因缺失和YTHDF2基因缺失都显著延长了GPX4 mRNA的半衰期(图4K)。因此,METTL14介导的GPX4 m6A修饰可通过YTHDF2依赖性识别促进mRNA降解。
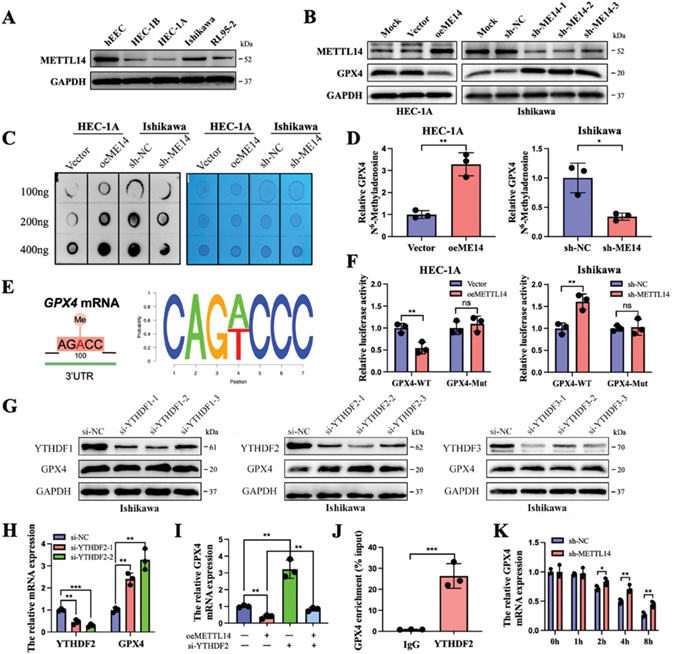
5. PRMT3表达与EC患者METTL14表达及预后良好呈负相关
在临床患者组织中进行了进一步的研究和证实。Western印迹结果显示,肿瘤组织中PRMT3的表达高于邻近的正常组织(图5A)。如图5B的代表性图像所示,肿瘤组织中PRMT3高表达,METTL14低表达(图5B)。值得注意的是,在PRMT3低表达的患者中,约有2/3(64.52%)的患者METTL14水平相对较高,而在PRMT3高表达组中,72.04%的组织METTL14水平较弱(图5C)。此外,Kaplan-Meier曲线显示,PRMT3表达较低的EC患者预后良好(图5D)。同样,qRT-PCR数据显示,新鲜EC肿瘤组织中的PRMT3 mRNA水平相对高于相应的邻近正常组织(图5E)。PRMT3的高表达与晚期病理分级、T分期、N分期和FIGO分期密切相关(图5F-I)。质谱鉴定出的与PRMT3相互作用蛋白在铁死亡途径中富集,进一步证实了作者的结论(图5J)。此外,作者还探讨了PRMT3、METTL14和GPX4联合作用的预后潜力。低PRMT3表达、高METTL14表达和低GPX4表达的患者预后最好。这些数据表明,由PRMT3、METTL14和GPX4组成的模型可用作EC患者的预后因素。总之,PRMT3在EC中表达上调,可能与肿瘤进展有关。
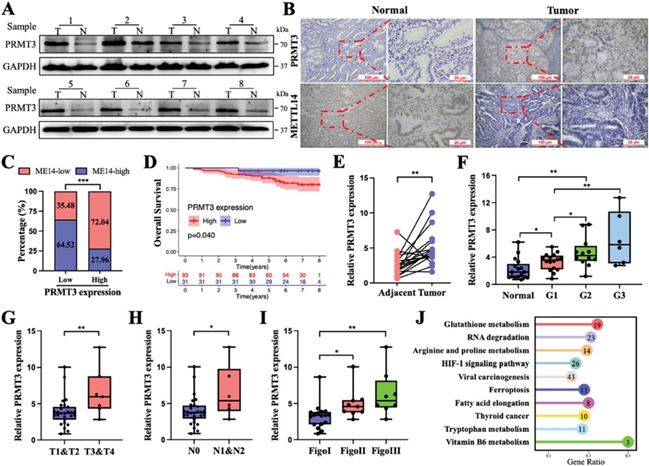
6. PRMT3抑制通过提高肿瘤对铁死亡的敏感性,增强了抗PD-1治疗对EC的抗肿瘤作用
从成年雌性NOG-dKO小鼠的尾静脉输入人外周血单核细胞(Hu-PBMCs),用于构建免疫重建模型。流式细胞术用于监测2-3周后小鼠外周血中人源T淋巴细胞的重建水平(图6A)。结果表明,人源化小鼠模型已成功建立,可用于后续的免疫检查点抑制剂研究。
作者在Hu-NOG-dKO小鼠模型中异种移植了HEC-1A-shPRMT3和HEC-1A-载体细胞,小鼠在注射后第11天开始在规定时间接受PD-1阻断或IgG治疗(图6B)。与其他小鼠相比,PRMT3下调和抗PD-1联合治疗组小鼠的肿瘤体积和重量进一步减少(图6C-E)。联合处理组的脂质过氧化物、MDA和Fe2+水平明显更高。Liproxstatin-1逆转了联合治疗组的疗效和细胞死亡程度(图6F-H)。此外,还观察到联合治疗组小鼠的GPX4表达最弱,4-HNE(一种脂质过氧化标记物)表达最强,PRMT3表达较低,METTL14表达较高(图6I)。PRMT3下调的癌细胞更容易被T细胞杀死(图6J)。与其他组相比,联合治疗组的肿瘤生长速度更慢,肿瘤重量更轻,脂质过氧化反应、MDA和Fe2+水平更高(图6K-N)。因此,阻断PRMT3表达或PRMT3抑制剂可能会通过抑制细胞对铁死亡的耐药性来提高抗PD-1疗法的疗效。
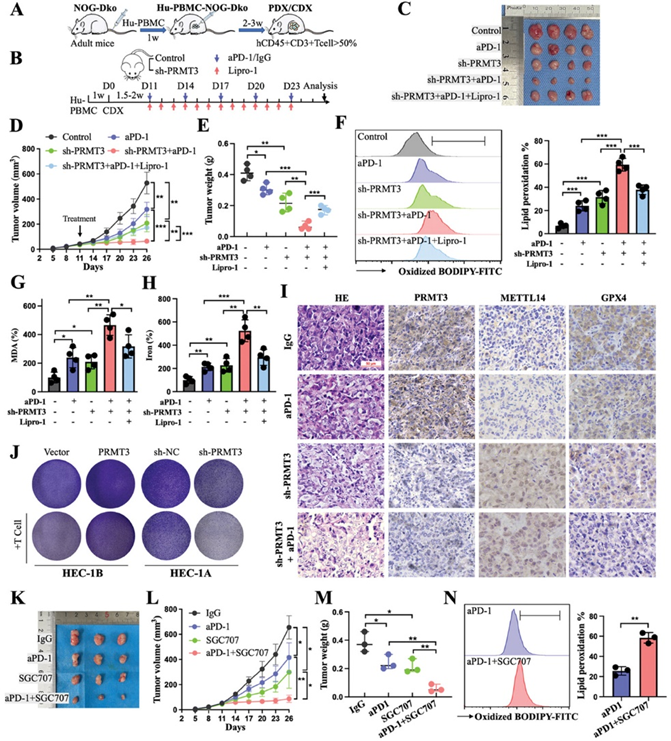
7. PRMT3下调增强EC放化疗的抗肿瘤疗效
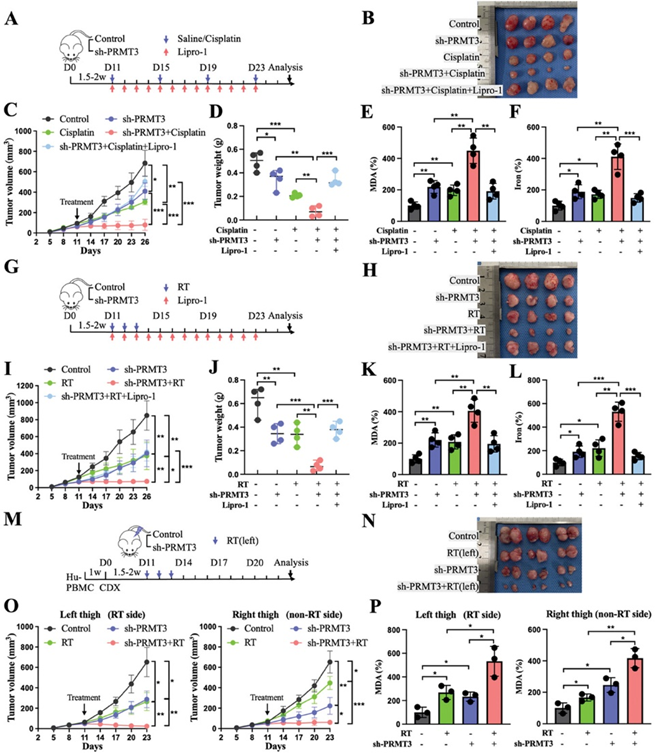
考虑到PRMT3基因敲除在增强免疫疗法方面的显著疗效,作者随后研究了它是否会对化疗和放疗等其他治疗方式产生类似的影响。事实证明,顺铂和放射治疗都会在细胞水平上加速铁死亡。此外,与其他对照组相比,顺铂和放射治疗在PRMT3缺陷组中都能产生更大的铁死亡诱导和治疗增敏作用,而且这种作用可被Liproxstatin-1部分消除(图7A-L)。鉴于肿瘤控制机制存在于辐射部位之外,作者进一步探讨了沉默PRMT3与放疗联合治疗是否有可能产生更大的远隔效应。在双侧肿瘤模型中,与未治疗的对照侧相比,治疗侧的肿瘤生长明显减慢,未治疗侧的肿瘤体积略有缩小,两侧的铁死亡水平均升高。与其他三组相比,双侧 PRMT3基因敲除肿瘤联合左侧放疗组的双侧肿瘤显示出更强的抗肿瘤效果和更大的铁死亡(图7M-P)。这些数据证实,阻断PRMT3改善了顺铂和放疗对肿瘤的抑制作用,并能更大程度地激活脱落效应。
结论:
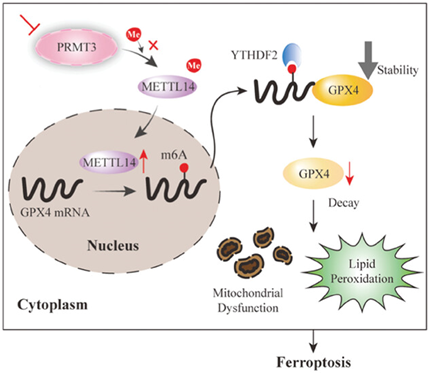
总之,作者发现PRMT3可能是预测EC患者不良预后和不利临床进展的生物标记物。作者还阐明了PRMT3在EC细胞铁死亡抵抗中的潜在机制。PRMT3缺失导致METTL14结合和精氨酸甲基化失败,METTL14高表达并以m6A依赖性方式下调GPX4,最终抑制细胞对铁死亡的抗性并导致EC的治疗敏感性(图8)。作者的工作提出了一种很有前景的方法,即消耗PRMT3可通过促进EC中的铁死亡抑制对抗PD-1、化疗和放疗的耐药性。
实验方法:
生物信息分析、蛋白质印迹分析、实时荧光定量PCR、铁检测、免疫共沉淀测定、m6 斑点印迹测定、MeRIP-qPCR、荧光素酶报告基因检测、RNA稳定性测定、免疫组化、转染、CCK8检测、菌落形成检测、脂质过氧化评估和细胞死亡分析、透射电子显微镜成像、免疫荧光、流式细胞术、T 细胞介导的癌细胞杀伤、Hu-NOG-dKO 模型的建立和体内疗效实验、建立 PDX
参考文献:
Wang, Y., Wang, C., Guan, X., Ma, Y., Zhang, S., Li, F., Yin, Y., Sun, Z., Chen, X., Yin, H., PRMT3-Mediated Arginine Methylation of METTL14 Promotes Malignant Progression and Treatment Resistance in Endometrial Carcinoma. Adv. Sci. 2023, 2303812.