






巨噬细胞表达ID1支持结肠癌细胞的干细胞性并限制CD8+ T细胞的浸润
消除肿瘤干细胞(CSCs)和恢复抗肿瘤免疫仍然是癌症治疗中未遇到的挑战。肿瘤相关巨噬细胞(tumor -associated macrophages, TAM)是肿瘤组织中重要的免疫细胞群,有助于形成CSC壁龛和抑制性免疫微环境。在这里,作者报告了TAMs中分化抑制因子1 (ID1)的高表达与结直肠癌(CRC)患者的不良预后相关。表达ID1的巨噬细胞维持肿瘤的干细胞性,抑制CD8+ T细胞的浸润。在机制上,ID1与STAT1相互作用诱导其细胞质分布,并抑制STAT1介导的SerpinB2和CCL4转录,这两种分泌因子负责肿瘤干细胞抑制和CD8+ T细胞募集。减少ID1表达可改善结直肠癌的进展,增强肿瘤对免疫治疗和化疗的敏感性。总之,作者的研究强调了ID1在控制TAM的肿瘤表型中的关键作用,并为ID1在CRC中的治疗靶向铺平了道路。本文于2023年11月发布于《Nature Communications》,IF=16.6。
技术路线:

主要研究结果:
1、TAMs中ID1的表达增强与结直肠癌患者的不良临床结果相关
一项基于转录组的研究表明,来自肿瘤组织的TAM在转录上与单核细胞及其各自的组织巨噬细胞不同。为了探索CRC TAMs中差异表达的致癌转录因子,作者深入挖掘了CRC细胞衍生条件培养基(CM)刺激下的腹膜巨噬细胞(PM)基因微阵列数据集(GSE80065)。在TAMs (CRC CM刺激的PM)中,与未刺激的PM (Ctrl)相比,2830个基因(log2FC > 1.5, P≤0.05)上调,其中80个基因编码转录因子,其中10个基因在CRC中表现出致癌特性(图1a)。在CT26或mc38来源的CM处理的RAW 264.7细胞中,验证了前5个基因的蛋白丰度。与对照组(Ctrl)相比,c- myc、Snai1和ID1,而不是Tcf4和ID2,在结肠癌细胞来源的cm处理组中被发现高表达(图1b, c)。由于c- myc和Snai1已被证明是替代巨噬细胞激活的关键参与者,作者随后关注ID1及其在TAM中的作用。与正常结肠组织中浸润的巨噬细胞相比,AOM/ dss诱导的结直肠癌模型或ApcMin自发结直肠癌模型中浸润的TAM均具有更高的ID1(图1d-f)。此外,从mc38衍生的同源原位肿瘤中分离的TAMs比从C57BL/6J小鼠中分离的pmms具有更高的ID1 mRNA和蛋白质表达(图1g, h)。作者还通过包含101例CRC患者标本的组织微阵列分析了人CRC TAMs中ID1的表达。在邻近正常组织中,CD68+ TAM中ID1的表达高于巨噬细胞(图1i, j)。作者还评估了在结直肠癌发展过程中TAM中ID1表达的潜在动力学改变。ID1在结直肠癌淋巴结转移患者的TAM中高表达,且与结直肠癌组织学分级和TNM分期呈正相关。此外,CD68+ TAM中ID1的高表达与结直肠癌患者预后不良相关(图1k)。总的来说,作者的研究结果表明,ID1在结直肠癌TAMs中异常高表达,尤其是晚期结直肠癌,预示着不良的临床预后。
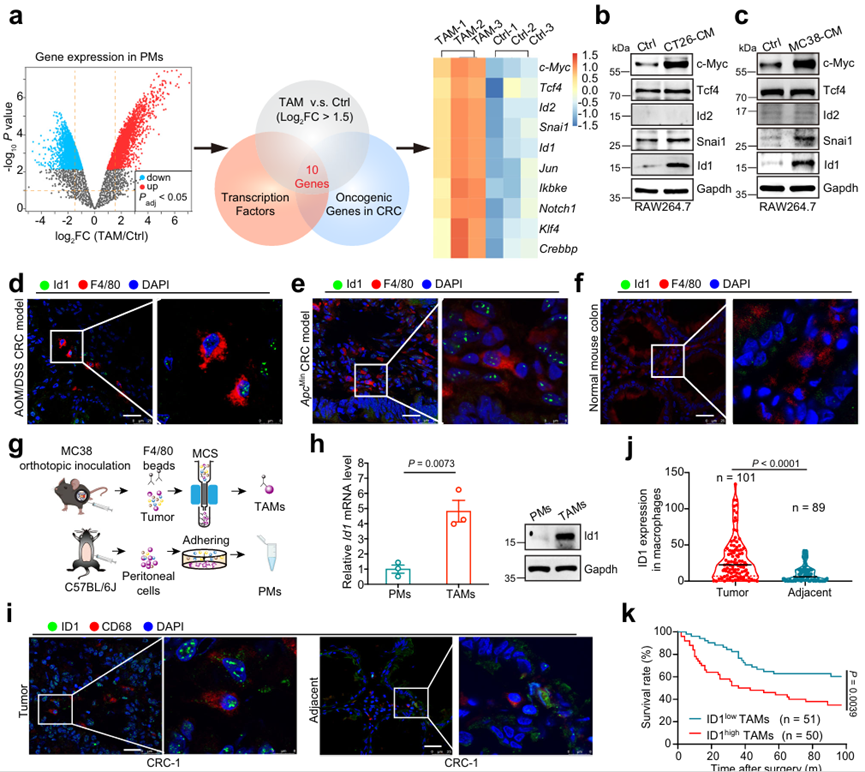
图1、TAMs中ID1的表达增强与结直肠癌患者的不良临床预后相关
2、表达TAMs的ID1促进结直肠癌的生长和转移
为了确定ID1是否直接调节巨噬细胞表型,作者在骨髓特异性基因启动子(Lyz2tm1(Cre)Ifo/J)的控制下,通过将含有ID1外显子的小鼠(ID1f/ f)与表达Cre重组酶的小鼠杂交,建立了骨髓特异性ID1缺陷小鼠(ID1Lyz-KO)。小鼠MC38结肠癌细胞皮下接种于ID1f/f和ID1Lyz-KO小鼠。与ID1f/f小鼠相比,MC38荷瘤ID1Lyz-KO小鼠的肿瘤生长降低,这与全体性ID1KO小鼠的数据一致。数据表明,表达ID1的骨髓细胞促进结直肠癌的进展。作者使用两种TAM过继转移模型进一步研究巨噬细胞ID1消融是否抑制肿瘤进展。首先,在体外用具有抗炎条件的基础培养基(白细胞介素-4)和巨噬细胞集落刺激因子(m-CSF))刺激骨髓源性巨噬细胞,诱导TAM表型。将上述TAM与MC38细胞混合,并过继转移至C57BL/6J受体小鼠。ID1f/f衍生的TAM加速了MC38细胞的肿瘤生长,而ID1的缺失减弱了TAM的促肿瘤作用。其次,分选从ID1f/f和ID1Lyz-KO小鼠的MC38源性肿瘤中分离出TAM,与MC38细胞混合,并过继转移到C57BL/ 6J受体小鼠体内(图2a)。含有ID1Lyz-KO TAM的肿瘤比含有ID1f/f TAM的肿瘤生长慢得多(图2b, c)。ID1Lyz-KO TAM组肿瘤结节中Ki67+细胞比例较低(图2d)。这些数据表明,TAMs中ID1的缺失会抑制结直肠癌的生长。TAM常通过影响分泌因子的产生,与肿瘤细胞或TME中的其他免疫细胞发生串扰。MC38荷瘤小鼠瘤内注射ID1f/f或ID1Lyz-KO TAMs衍生的CM。注射来自ID1Lyz-KO TAM的CM的肿瘤生长速度比注射来自ID1f/f TAM的CM的肿瘤生长速度慢。为了研究巨噬细胞中的ID1是否参与肿瘤转移的控制,作者建立了3种CRC转移模型。在脾-肝转移模型中,将荧光素酶标记的CT26细胞与TAM来源的CM腹腔注射一起注入脾内(图2e)。注射ID1f/f TAMs衍生cm的小鼠显示出增强的生物发光信号、肝/体重比、肿瘤直径和转移性肿瘤结节的肝叶数量,当在TAMs中消融ID1时,这些都是相反的(图2f-k)。在CT26原位肝转移模型中,将高度转移的CT26细胞注射到盲肠壁的浆膜下层,并通过腹腔注射TAM来源的CM(图2l)。用ID1Lyz-KO TAM衍生的cm治疗组,转移部位更少,转移瘤直径更小(图2m - o)。在结直肠癌肺转移模型中,通过尾静脉注射荧光素酶标记的CT26细胞,并通过腹腔注射TAM来源的CM。ID1Lyz-KO TAM衍生cm治疗组肺转移面积更小,肺/体重比更低。这些数据表明,表达ID1的TAM通过影响其分泌成分促进结直肠癌肿瘤的生长和转移。据报道,促炎刺激诱导巨噬细胞具有抗肿瘤(m1样)表型。IFN-γ和LPS刺激RAW 264.7细胞(如图RAW- ctrl所示)和ID1过表达的RAW 264.7细胞(如图RAW- ID1oe所示),诱导肿瘤抑制表型(图2p)。作者发现来自RAW-Ctrl细胞的CM减轻了肿瘤细胞的生长,降低了Ki67+细胞的比例,而ID1异位表达逆转了这些抑制作用(图2q, r)。这些数据表明,高ID1表达损害了经典活化巨噬细胞的抗肿瘤表型。
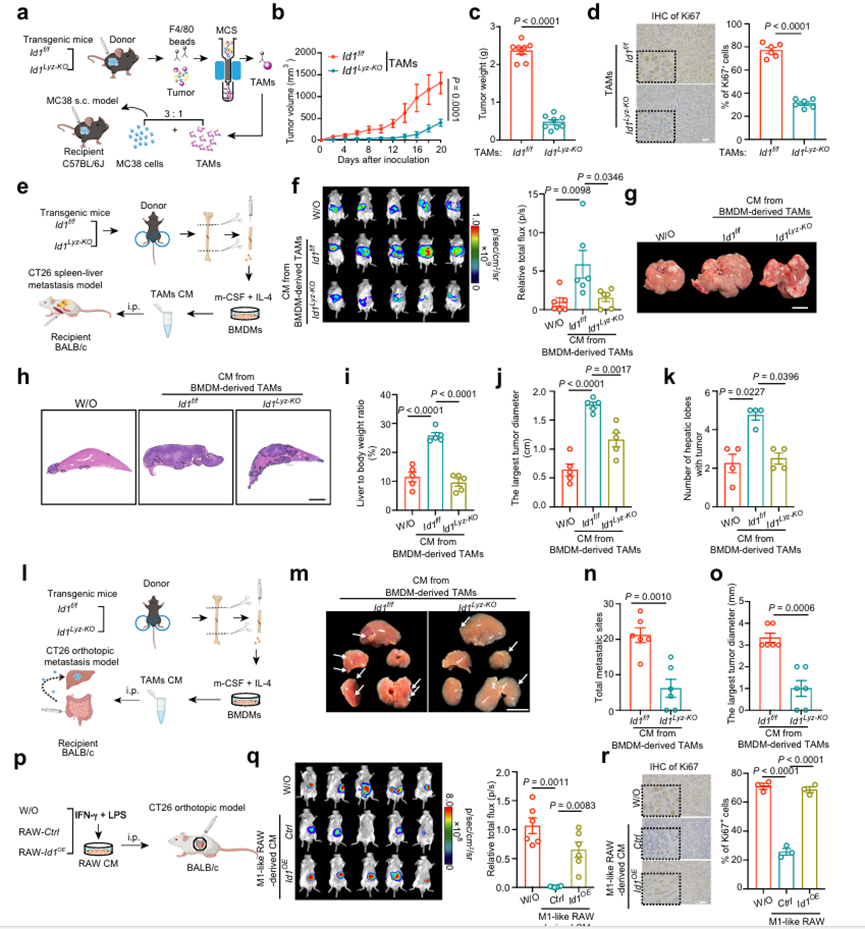
图2、表达TAM促进结直肠癌的生长和转移
3、表达TAMs的ID1部分通过排除CD8+ T细胞募集来促进结直肠癌的发展
TAM通常通过防止肿瘤细胞受到T细胞的攻击而具有直接的免疫抑制功能。为了评估表达ID1的TAM是否具有免疫抑制功能,如图2a所示,将MC38细胞和TAM混合接种于免疫缺陷的BALB/c裸鼠,而不是免疫正常的C57BL/6J小鼠。BALB/c裸小鼠的肿瘤抑制率约为50%,而C57BL/6J小鼠(如图2a所示)的肿瘤抑制率超过80%(图3a),表明表达ID1的TAM可能破坏T细胞介导的抗肿瘤免疫应答。TAMs中ID1的缺失增加了免疫活性C57BL/6J小鼠mc38衍生肿瘤中CD8+ T细胞的浸润(图3b, c)。此外,TAMs中ID1的缺失增加了肿瘤浸润性CD8+ T细胞中干扰素-γ (IFN-γ)和颗粒酶B的表达(图3d)。作者还利用BALB/c裸鼠脾-肝转移模型评估了ID1在抗肿瘤免疫应答中的作用。与ID1f/f TAM中的CM相比,ID1Lyz-KO TAM中的CM可减轻BALB/c裸小鼠的生物发光信号、肝/体重比和肿瘤直径。然而,在免疫正常小鼠中,ID1缺失导致的TAM肿瘤抑制率从70%下降到40%,在免疫缺陷小鼠中(图3e),证实了ID1表达的TAM调节T细胞介导的抗肿瘤免疫。用来自ID1f/f TAM的CM治疗可减少肝或肺转移瘤内区域的CD8+ T细胞浸润(如图2e、l),通过敲除TAM中的ID1可逆转这一情况(图3f-h)。此外,在原位结直肠癌模型中,用来自m1样RAW 264.7细胞的CM处理(如图2p所示)增加了肿瘤中CD8+ T细胞的浸润,这被ID1过表达所消除(图3i)。为了验证CD8+ T细胞参与表达ID1的TAM的肿瘤促进作用,作者在CT26 s.c.模型中使用全身给药抗CD8β抗体来耗尽CD8+ T细胞(图3j)。与ID1Lyz-KO TAMs的CM治疗相比,ID1Lyz-KO TAMs治疗降低了肿瘤生长,这被CD8+ T细胞消耗部分逆转(图3k-m),肿瘤抑制率从~85% (IgG治疗组)到35% (CD8消耗抗体组)(图3n)。这些数据表明,表达ID1的TAM的促肿瘤作用依赖于CD8+ T细胞。缺乏肿瘤内T细胞浸润与T细胞增殖或迁移减少有关。m1样RAW 264.7细胞中的ID1OE抑制CD8+ T细胞迁移,而TAM中的ID1Lyz-KO则增强CD8+ T细胞迁移(图3o,p)。作者还评估了ID1操纵巨噬细胞对T细胞增殖和杀伤活性的影响。用羧基荧光素琥珀酰酰酯(CFSE)荧光染料标记CD8+ T细胞,作者发现在m1样RAW 264.7细胞中过表达ID1或在TAM中耗尽ID1对CD8+ T细胞的增殖没有影响。此外,ID1f/f组和ID1Lyz-KO组在影响ova特异性CD8+细胞毒性OT-1 T细胞杀伤活性方面没有差异。作者也证实了作者在人类原代细胞共培养系统中的观察结果。当与CRC患者外周血单个核细胞(PBMCs)分离的CD8+ T细胞共培养时,与对照组相比,CRC患者肿瘤组织分离的TAM中ID1的缺失显示CD8+ T细胞迁移率增强(图3q)。ID1表达的MDSCs有促进肿瘤生长的报道,作者进一步探讨它们是否也通过影响CD8+ T细胞浸润发挥促瘤作用。从ID1f/f和ID1Lyz-KO小鼠mc38来源肿瘤中分离的Cd11b+ Gr1+ MDSCs与CD8+ T细胞共培养。作者发现MDSCs中ID1的缺失对CD8+ T细胞的迁移速率没有影响。这些数据表明,表达TAMs而非MDSCs的ID1阻碍CD8+ T细胞募集并促进肿瘤细胞逃避免疫消除。
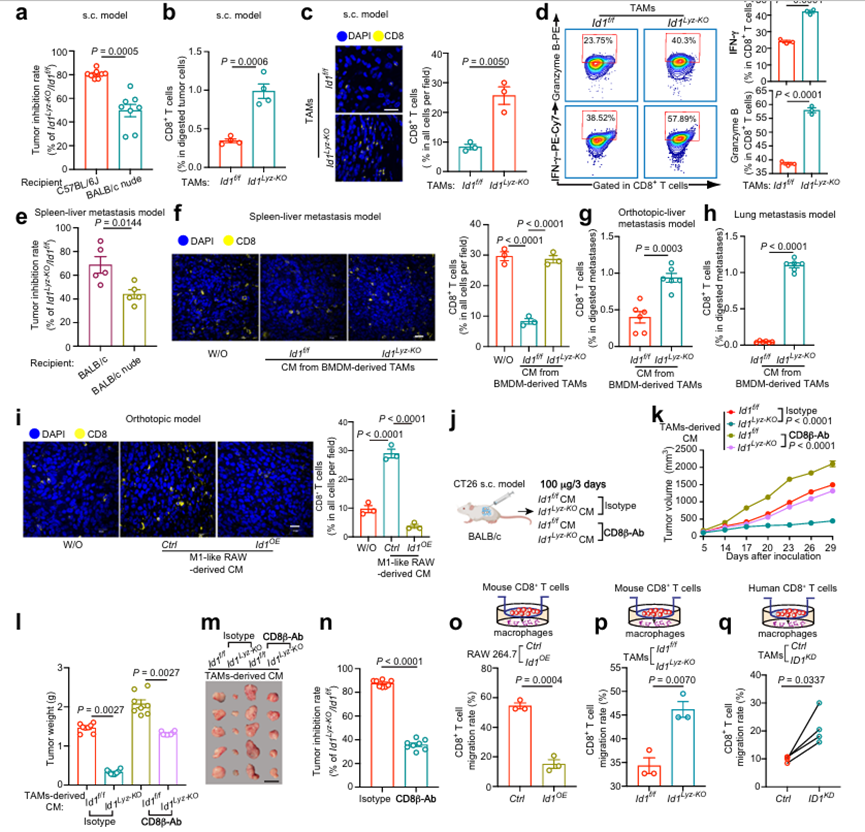
图3、表达ID1的TAM部分通过抑制CD8+ T细胞募集促进结直肠癌进展
4、表达ID1的TAM是通过激活FAK-YAP信号通路来维持CRC干性性状的必要条件
敲除TAMs中的ID1对免疫缺陷小鼠的肿瘤生长和转移有抑制作用,表明表达ID1的TAMs可能影响肿瘤的内在特性。CSC被认为是肿瘤发生、转移、耐药和复发的起源。如上所述,ID1Lyz-KO组分离的肿瘤细胞表达CD44和Lgr5 (CSC标志物)低于ID1f/f组(图4a, b)。TAM中ID1的缺失使肿瘤启动细胞频率降低到近1/3(从1/979降至1/ 2378)(图4c)。这些数据表明,表达TAMs的ID1支持结直肠癌细胞的干性特征。TAM可以通过可溶性介质促进CSC表型。与CRC患者肿瘤组织来源的ID1KD TAM共培养的HCT116细胞表达的CD44和LGR5水平低于与Ctrl TAM共培养的细胞(图4d)。与ID1Lyz-KO TAM共培养的MC38细胞表达的CD44和Aldh水平低于与ID1f/f TAM共培养的MC38细胞(图4e)。CT26细胞与过表达ID1的m1样RAW 264.7细胞共培养,HCT116细胞与过表达ID1的m1样THP-1细胞共培养,CD44和ALDH的表达高于与Ctrl细胞共培养的细胞。此外,来自ID1KDor ID1Lyz-KO TAMs的CM被抑制,而来自ID1OE RAW 264.7细胞或ID1OE THP-1细胞的CM促进了肿瘤球的形成能力(图4f;)和CRC细胞的侵袭性。这些数据表明,TAMs中的ID1通过改变TAMs的分泌成分来增加CRC细胞的干性特征。通过混合MC38细胞和ID1f/f/ID1Lyz-KO TAMs植入sc肿瘤结节,利用转录组测序技术评估从肿瘤结节分离的癌细胞中的差异表达基因(DEGs)(图4)。基因集富集分析(GSEA)表明,TAMs中atID1的缺失导致局灶黏附激酶(FAK)和ye相关蛋白(YAP)信号通路的抑制,但不影响其他肿瘤干性相关信号通路,如Notch、Wnt/βcatenin和Sonic hedgehog (SHH)通路(图4h, 1)。与该数据一致的是,从ID1f/f小鼠的肿瘤结节中分离出的肿瘤细胞与BMDM衍生的TAM混合后,与未混合的细胞相比,p-Fak和Yap的丰度更高,这是通过TAM中ID1的消耗而逆转的(图4j)。作者还在MC38 s.c.模型(如图2a所示)、脾-肝转移模型(如图2e所示)、原位模型(如图2p所示)以及体外细胞非接触共培养系统中检测到了FAK-YAP信号的变化。ID1缺失的TAMs减少,而ID1过表达的m1样RAW 264.7细胞增加了p-Fak和Yap的丰度。FAK特异性抑制剂Y15可以逆转m1样THP1细胞中ID1过表达对YAP蛋白表达的上调作用,表明ID1通过FAK依赖的方式诱导YAP活化(图4k)。FAK通过磷酸化YAP的Y357位点来维持肿瘤干性性状,从而增强其蛋白稳定性、核易位和转录活性。YAP在核易位后与TEAD形成转录复合物,是维持肿瘤干性性状的关键步骤。ID1缺失下调,而其过表达增强了YAP蛋白的稳定性、YAP (Y357)磷酸化、核易位、TEAD的荧光素酶活性以及下游靶基因的转录(图41 - O)。维替波芬是YAP-TEAD复合物的一种抑制因子,在很大程度上逆转了ID1过表达m1样THP-1细胞的CM对CRC肿瘤干细胞的促进作用(图4p)。这些数据表明,来自表达TAMs的ID1分泌成分对于维持肿瘤细胞中FAK-YAP的激活至关重要。Y15进一步验证表达ID1的TAM是否通过激活FAK信号维持癌干性性状。在CT26 s.c. BALB/c裸鼠模型中,与对照组相比,Y15如预期的那样缓解了肿瘤生长,逆转了ID1OE RAW 264.7细胞中CM的肿瘤加速作用(图4q, r)。值得注意的是,在Y15处理下,ID1OE m1样巨噬细胞中的CM不再增强肿瘤细胞的侵袭性、肿瘤球体形成能力或CD44蛋白的丰富度(图4s)。这些数据表明,表达ID1的TAM通过激活癌细胞中的FAK-YAP信号来支持CRC的干细胞性。
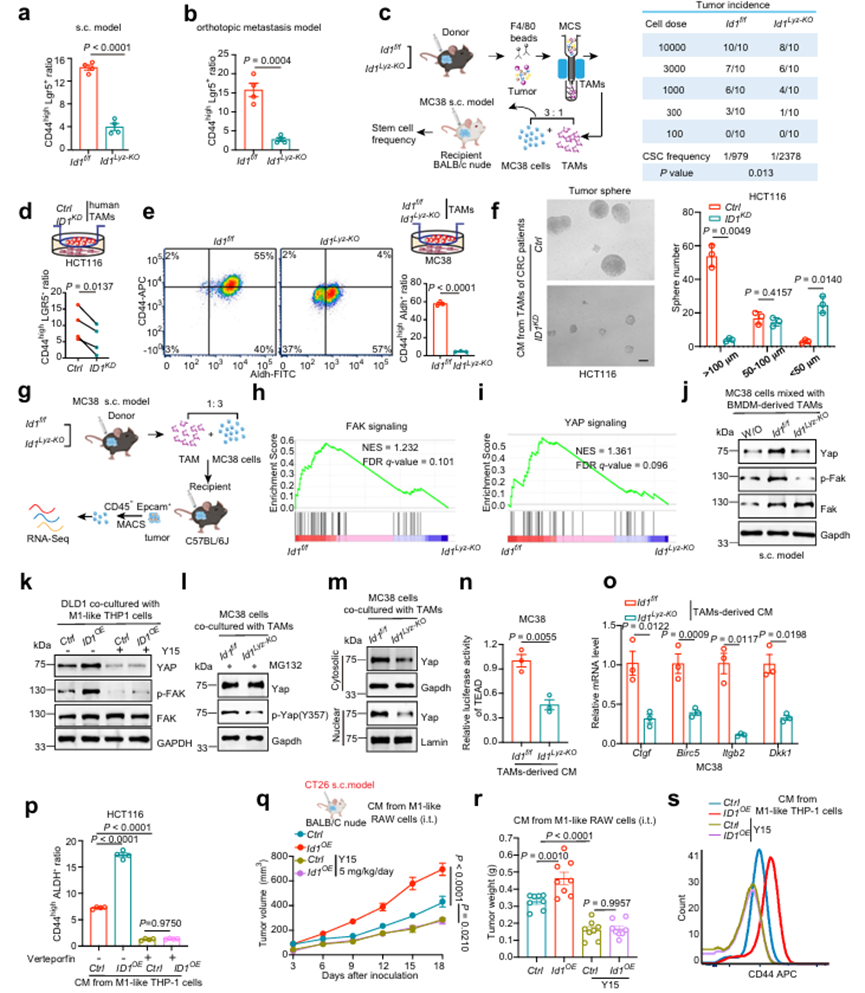
图4、表达ID1的TAM对于通过激活FAK-YAP信号通路维持CRC干性性状至关重要
5、TAMs中的ID1通过抑制CCL4和SerpinB2转录介导肿瘤免疫逃避和CRC干性维持
对ID1f/ ID1Lyz-KO小鼠中mc38来源的肿瘤组织分离的TAM进行转录组测序,以探索ID1如何塑造TAM的促肿瘤表型(图5a)。ID1抑制与其相互作用的bHLH蛋白的DNA结合和转录激活能力。作者的上述数据表明,ID1调节的分泌成分对TAM维持癌症的干细胞性和促进癌症免疫侵袭至关重要。因此,作者将重点放在ID1Lyz-KO TAM中编码分泌蛋白的基因与ID1f/f TAM相比的上调上。在ID1Lyz-KO TAMs中,与ID1f/f TAMs相比,发现超过2000个基因增加(图5b)。从免疫应答调节方面,作者选择了20个log2FC > 1.5的上调基因编码趋化因子,其中Ccl3、Ccl4、Cxcl9和Cxcl16具有共同的抑瘤作用和促进T细胞浸润的作用(图5c)。在这四种趋化因子中,只有Ccl4在ID1OE RAW 264.7细胞中的转录水平低于Ctrl细胞(图5d)。酶联免疫吸附试验(ELISA)证实,从结直肠癌患者肿瘤组织、mc38衍生肿瘤组织或BMDMs诱导的ID1缺失的TAM显示CCL4蛋白丰度增加(图5e)。m1样巨噬细胞中ID1的过表达降低了CCL4蛋白水平(图5f)。然后,作者建立了Ccl4缺失的TAM来验证Ccl4在介导ID1调节的CD8+ T细胞募集中的作用。TAM中ID1的缺失导致CD8+ T细胞迁移增加,这可以通过敲低Ccl4来逆转(图5)。这些数据表明,表达ID1的TAM失去了CCL4的分泌,阻碍了CD8+ T细胞向肿瘤部位的运输。从肿瘤内在特性调控方面,作者选择了12个log2FC > 1.5的编码分泌性肿瘤抑制因子(不含趋化因子)的上调基因,其中只有Serpin家族B成员2 (SerpinB2)被报道抑制FAK信号传导(图5h)。分析CRC GEO数据集,作者发现SerpinB2与CRC患者的满意结局呈正相关。TAMs中ID1的缺失增加了SerpinB2蛋白的丰度,而ID1过表达降低了SerpinB2蛋白的丰度(图5i, j)。TAMs中ID1敲除对CSC标志物表达、肿瘤球形成能力和肿瘤侵袭性的抑制作用可以通过去除Serpinb2 (Serpinb2KD)来逆转(图5 k、m)。此外,在TAM中沉默Serpinb2可以增强肿瘤侵袭性和肿瘤成球能力,这可以通过Y15治疗逆转。这些数据表明,ID1可以抑制Serpinb2的表达,激活FAK信号,增强癌细胞的干细胞性。为了证实ID1是否通过损害SerpinB2和CCL4的表达而发挥促瘤作用,作者在ID1f/f和ID1Lyz-KO TAM中同时缺失了这两个基因。当Ccl4和Serpinb2同时缺失时,TAM中ID1缺失对肿瘤生长和肝转移的抑制作用被完全消除(图5n-p)。无论是在s.c.模型中还是在转移模型中,当Ccl4和Serpinb2同时被敲低时,TAMs中ID1缺失引起的CD8+ T细胞浸润增强和对FAK-YAP信号的抑制都减弱了。证据表明,ID1抑制巨噬细胞SerpinB2和CCL4的转录,促进肿瘤免疫逃逸,增强CRC细胞的致瘤能力,最终导致肿瘤生长和转移加剧。
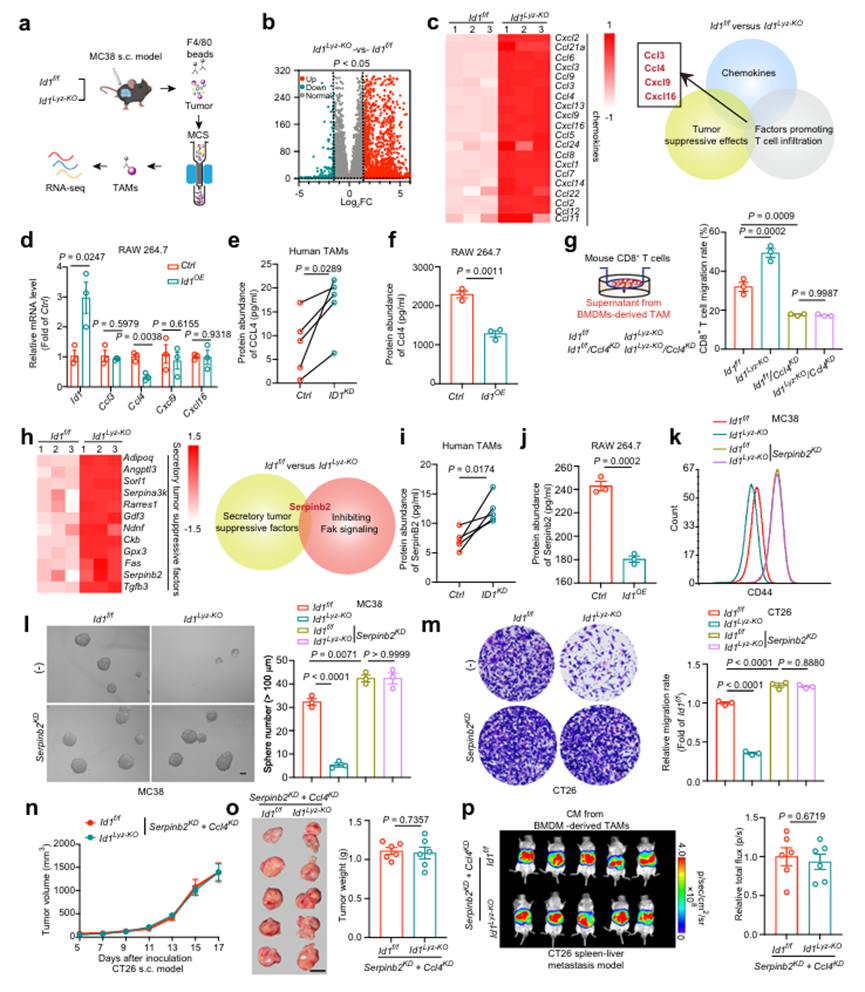
图5、TAMs中的ID1通过抑制CCL4和SerpinB2转录介导肿瘤免疫逃避和CRC干性维持
6、ID1与STAT1相互作用抑制CCL4和SerpinB2的转录
由于ID1不具有DNA结合能力,通常与TF相互作用并抑制其活性,因此采用共免疫沉淀联用质谱法(Co-IP/MS)鉴定RAW 264.7细胞中可能与ID1相互作用的TF。此外,使用hTFtarget工具预测了16个可能负责CCL4和SerpinB2转录的TF。根据Co-IP/MS分析,STAT1是16个TF中唯一一个与ID1相互作用的TF(图6a)。作者通过Co-IP、近距离结扎实验(PLA)和共聚焦成像进一步证实了CRC患者肿瘤组织中HEK 293T细胞、RAW 264.7细胞和CD68+ TAM中STAT1和ID1的直接相互作用(图6b-e)。这些数据表明ID1可能与STAT1相互作用,抑制STAT1介导的CCL4和SerpinB2转录。采用染色质免疫沉淀法(ChIP)验证了Serpinb2和Ccl4是否为RAW 264.7细胞中Stat1的靶基因。Stat1在−3000 ~−2701 bp、−2760 ~−2461 bp和−1080 ~−781 bp的位置与Ccl4启动子结合;以及在- 2517至- 2221 bp和- 1320至- 1021 bp的Serpinb2启动子。为了进一步验证,作者选择了结合信号最高的启动子区域pCcl4-2和pSerpinb2-8。双荧光素酶报告基因实验表明,ID1通过结合pCcl4-2和pSerpinb2-8启动子区域抑制Ccl4和Serpinb2的转录(图6f)。为了进一步探索ID1调控STAT1转录活性的分子机制,作者深入剖析了这两种蛋白的结合结构域。Co-IP分析表明,ID1的n端和HLH结构域介导了其与STAT1的相互作用。此外,STAT1的n端、DNA结合域和SH2域介导了其与ID1的关联。据报道,n端和SH2结构域介导STAT1二聚化。然而,ID1过表达对STAT1二聚化没有影响。STAT1的细胞质和细胞核定位是一个维持适度信号激活的动态过程。因此,作者质疑ID1是否参与了STAT1核出口的调控。在IFN-γ处理下,ID1的过表达降低了细胞核中基础Stat1和p-Stat1的丰度,但增加了细胞质中的Stat1和p-Stat1的丰度(图6g)。CRM1识别STAT1 DNA结合域的一个区域,将其重新分配回细胞质。作者发现ID1增强了CRM1与STAT1的相互作用(图6h, i)。Co-IP分析进一步证实了这三种蛋白相互作用,这意味着在ID1、STAT1和CRM1之间形成了异三聚体蛋白复合物。Leptomycin B是一种CRM1抑制剂,可保持Stat1的核分布。在IFN-γ处理下,ID1将STAT1二聚体从细胞核重新分配到细胞质中,这可以通过leptomycin B处理消除(图6j)。此外,作者发现在leptomycin b处理下,ID1缺失并不影响Stat1与Serpinb2和Ccl4启动子的结合能力。这些数据表明,ID1促进CRM1向STAT1募集,从而促进STAT1的细胞质分布,抑制STAT1诱导的CCL4和SerpinB2转录
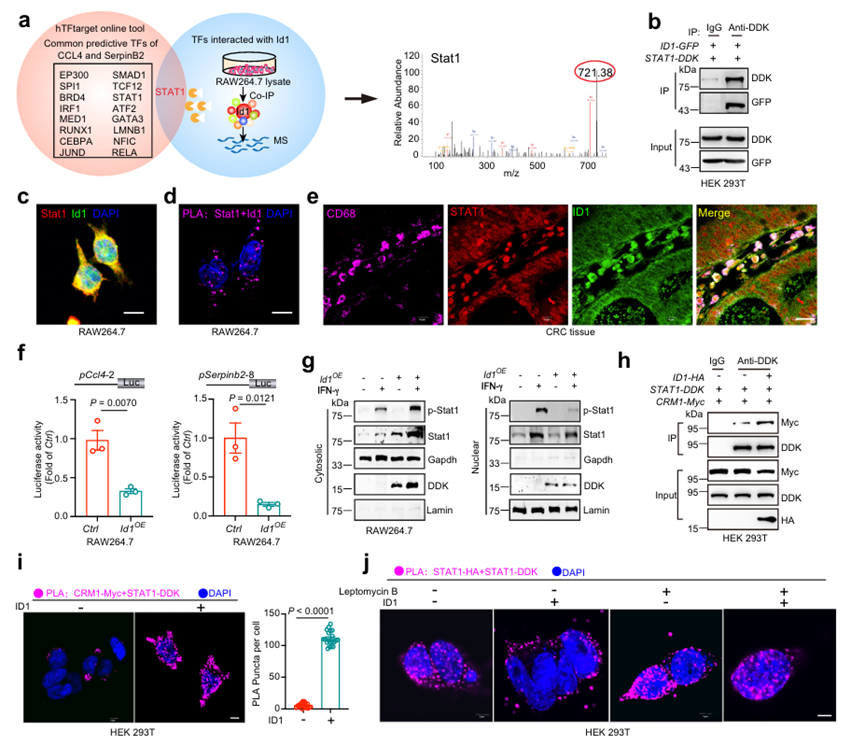
图6、ID1与STAT1相互作用抑制CCL4和SerpinB2的转录
7、靶向ID1抑制结直肠癌进展并使肿瘤细胞对化疗和免疫治疗敏感
ID1是一种短寿命蛋白,主要受到泛素化介导的蛋白酶体或自噬降解的影响。作者探讨了药物减少ID1是否可以消除TAM的促肿瘤作用。泛素化特异性蛋白酶1 (USP1)是一种去泛素化酶,据报道可以保持蛋白的稳定。作者评估了ML323 (USP1的选择性抑制剂)在同基因sc型结直肠癌小鼠模型和同基因原位转移型结直肠癌小鼠模型中的潜在抗肿瘤作用。在s.c.模型中,ML323给药降低了TAMs中ID1的表达,并表现出明显的肿瘤抑制作用,从肿瘤体积和肿瘤重量与载药组比较可见(图7a, b)。此外,作者检测到ml323处理组肿瘤浸润性CD8+ T细胞中颗粒酶B和IFN-γ表达增加,CD8+ T细胞效应功能增强(图7c)。此外,ml323处理组肿瘤组织中分离的肿瘤细胞中CSC标志物的表达降低(图7d)。在原位肝转移模型中,ML323治疗明显减轻了肝转移,CD8+ T细胞浸润和有效性增强,肿瘤细胞干性性状受到抑制(图7e-i)。作者进一步证实,ML323部分通过调节TAMs中ID1的表达来抑制肿瘤进展。使用ID1f/f和ID1LyzKO小鼠,作者证明在MC38 s.c.模型中,骨髓细胞中ID1的缺失在很大程度上但不是完全逆转了ML323的肿瘤抑制作用(图7j, k),表明ML323在髓细胞中部分通过ID1起作用。然后将TAM从肿瘤组织中分离出来,置于载具或ML323处理的小鼠中,以研究ML323的影响(图71)。通过非接触式共培养系统,作者证明了ML323处理的TAM促进了CD8+ T细胞的迁移,抑制了CT26细胞中CD44和Aldh的表达(图7m, n)。此外,体外实验进一步证实了ML323对TAM的直接影响。作者证实,BMDM衍生TAM的CM与ML323治疗也促进了CD8+ T细胞的迁移能力,降低了CSC标志物的表达,肿瘤成球能力和侵袭性。此外,TAMs中的ID1Lyz-KO消除了ML323治疗对肿瘤干细胞和CD8+ T细胞迁移的影响(图7o,p),表明ML323对TAM的影响是由ID1介导的。在机制上,在ML323处理的TAM中,Serpinb2和Ccl4的蛋白丰度均上调(如图7l所示)。作者使用RAW 264.7细胞进行体外实验,进一步验证ML323通过调节ID1对Serpinb2和Ccl4的影响。作者的研究结果表明,ML323处理导致ID1过表达阴性的RAW 264.7细胞中Serpinb2和Ccl4的表达增加(图7q, r)。这些数据共同表明,ML323通过降低TAMs中ID1的表达来发挥抗肿瘤作用。这些结果促使作者在临床前小鼠模型中评估ML323(较低剂量为3mg /kg)联合化疗或免疫治疗的潜在效果。首先,作者用ML323联合5-氟尿嘧啶(5-FU)治疗CT26荷瘤小鼠(图7s)。与5-FU或ML323单药治疗组相比,ML323联合5-FU表现出协同抗肿瘤作用,从肿瘤体积(图7t)和肿瘤重量(图7u, v)可见。其次,作者在CT26荷瘤小鼠中测定了ML323与抗ctla4的协同作用(图7w)。联合治疗的抗肿瘤效果强于单独使用ML323或抗ctla4(图7x-z)。此外,在联合治疗组中观察到CD8+ T细胞浸润增加,CD8+ T细胞效应功能增强。这些数据表明,通过抑制USP1活性靶向ID1可能是提高化疗和免疫治疗CRC疗效的潜在策略。由于所有的实验都是在雄性小鼠身上进行的,作者进一步研究ID1在TAM中的作用是否存在性别差异。在雄性小鼠中观察到,注射来自ID1Lyz-KO TAM的CM的肿瘤生长速度比注射来自ID1f/f TAM的CM的肿瘤生长速度慢。此外,ID1Lyz-KO组肿瘤组织中CD45- Epcam+肿瘤细胞CD8+ T细胞浸润增多,CD8+ T细胞活性提高,CD44high Lgr5+表达降低。这些数据表明,ID1在TAM中的促肿瘤作用在性别之间没有差异。为了探索作者的发现的普遍性,作者建立了另外两个使用H22(小鼠肝细胞癌细胞系)和Pan02(小鼠胰腺导管腺癌细胞系)的sc肿瘤小鼠模型。BMDM衍生TAM中ID1的缺失也能抑制肝癌和胰腺癌的肿瘤生长,降低肿瘤干细胞比例,促进CD8+ T细胞的浸润和活性。这些数据表明,作者关于表达TAM的ID1促进肿瘤进展的发现可能与其他类型的癌症具有广泛的相关性。
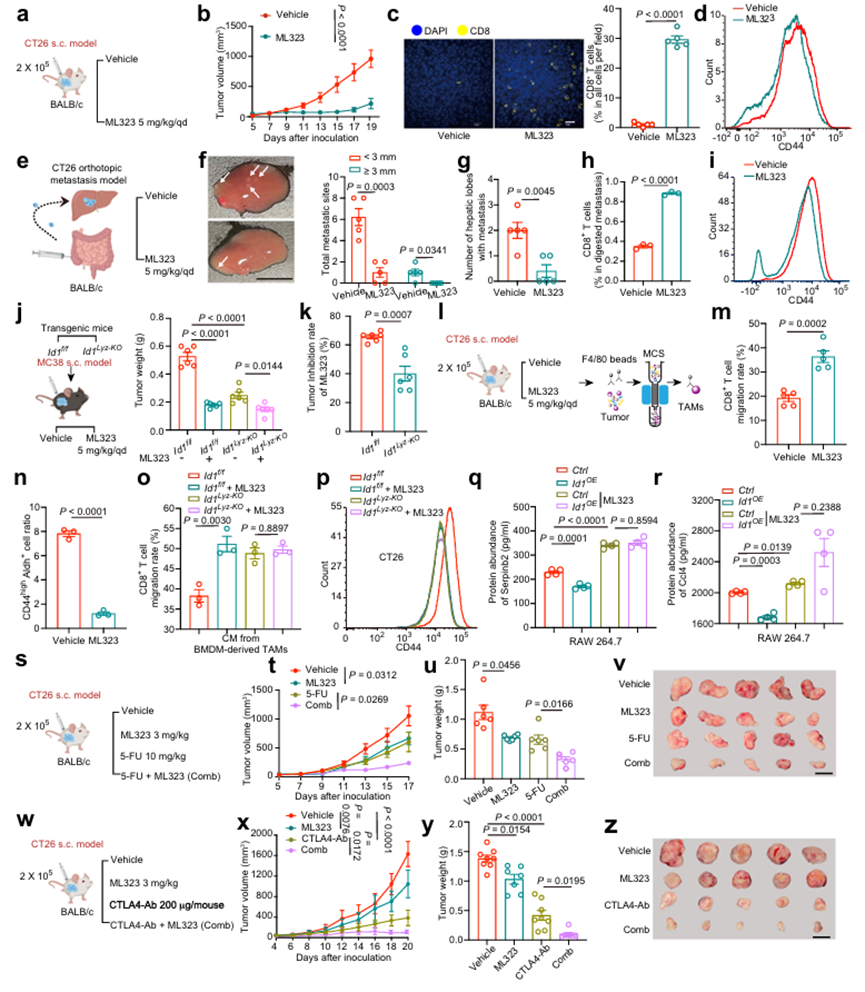
图7、靶向ID1抑制结直肠癌进展并使肿瘤细胞对化疗和免疫治疗敏感
结论
总的来说,作者的工作确定了TAM表达的ID1作为中心分子节点,双重控制癌症启动能力和诱导肿瘤免疫逃逸。在结直肠癌中,TAMs中ID1的高表达代表了一种免疫逃避和肿瘤干细胞维持机制。此外,作者提供的证据表明,靶向ID1稳定性可以提高CRC患者的免疫治疗和化疗敏感性。
实验方法
流式细胞术、质粒构建、免疫沉淀,免疫印迹,免疫染色和免疫组织化学、质谱分析、肿瘤球形成试验、羧基荧光素丁二酰酯(CFSE)测定
参考文献
Shang S, Yang C, Chen F, Xiang RS, Zhang H, Dai SY, Liu J, Lv XX, Zhang C, Liu XT, Zhang Q, Lu SB, Song JW, Yu JJ, Zhou JC, Zhang XW, Cui B, Li PP, Zhu ST, Zhang HZ, Hua F. ID1 expressing macrophages support cancer cell stemness and limit CD8+ T cell infiltration in colorectal cancer. Nat Commun. 2023 Nov 23;14(1):7661. doi: 10.1038/s41467-023-43548-w. PMID: 37996458; PMCID: PMC10667515.