






肝癌铁死亡新机制:靶向USP8通过稳定OGT抑制SLC7A11 O-GlcNAc糖基化
肝细胞癌(Hepatocellular Carcinoma,HCC)是原发性肝癌中最常见的亚型,是一种致命的侵袭性恶性肿瘤。泛素化是一种翻译后修饰,在细胞存活、细胞凋亡、细胞周期、抗原提呈等过程中发挥重要作用。去泛素化酶(Deubiquitylating Enzymes,DUBs)可以调控蛋白质的泛素化修饰,与多种癌基因和肿瘤相关蛋白的表达有关,因此DUB抑制剂的开发成为癌症治疗中有吸引力的靶点。本研究考察DUBs抑制剂在HCC中的抗肿瘤作用,发现泛素特异性肽酶8(Ubiquitin Specific Peptidase 8,USP8)小分子抑制剂DUB-IN-3有显著的抗癌反应。体内外实验证明靶向USP8参与铁死亡并抑制HCC增殖和肺转移。进一步机制研究表明,USP8去泛素化修饰O-GlcNAc糖基转移酶(O-GlcNAc Transferase,OGT)蛋白,提高其蛋白的稳定性,后者通过O-GlcNAc糖基化调控铁死亡关键蛋白SLC7A11的Ser26位点的糖基化,从而抑制肝癌铁死亡,促进肝癌进展。以上研究表明了USP8可能是一个潜在的HCC治疗靶点。
该研究于2023年10月发表在《Advanced Science》,IF:15.1
技术路线
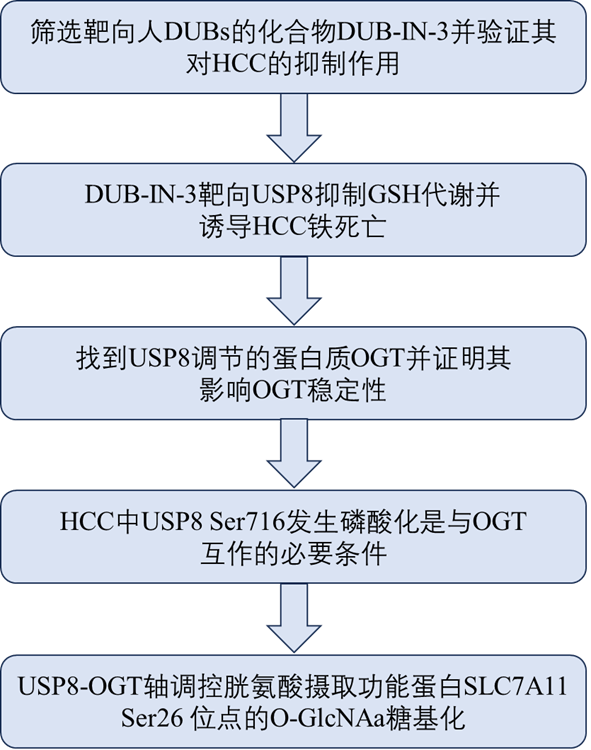
主要研究结果
1. DUB-IN-3有效抑制HCC
为了发现有效的HCC生长抑制剂,作者在6种HCC细胞系中筛选了23种已知靶向人DUBs的化合物。所有细胞系都对USP8抑制剂DUB-IN-3表现出高敏感性(图1A,B)。作者还发现DUB-IN-3以剂量和时间依赖性的方式降低了LM3和Hep3B细胞的细胞活力(图1C,D)。接下来通过PI染色测试了DUB-IN-3对细胞死亡的影响。如图1E所示,DUB-IN-3显著诱导HCC细胞死亡;Calcein AM/PI染色结果证明了这一结论(图1F,G)。作者进一步从原发性肝癌中建立6种患者来源的类器官,并测试DUB-IN-3的抗癌活性,发现DUB-IN-3在类器官模型中表现出显著的癌症抑制作用(图1H,I)。
为了进一步研究DUB-IN-3的抗癌效果,作者选择对DUB-IN-3处理有较高敏感性的LM3和Hep3B细胞系进行进一步分析。CCK8分析表明DUB-IN-3抑制HCC细胞的增殖(图2A)。克隆形成试验表明DUB-IN-3显著抑制HCC细胞的克隆形成能力(图2B)。EdU实验结果表明DUB-IN-3抑制LM3和Hep3B细胞的DNA合成(图2C)。Calcein AM/PI染色表明DUB-IN-3导致细胞死亡增加(图2D)。Transwell试验表明DUB-IN-3显著降低LM3和Hep3B细胞的侵袭能力(图2E)。接下来研究了DUB-IN-3在HCC干性特征中的作用,发现DUB-IN-3 显著抑制了LM3和Hep3B细胞的成球能力(图2F)。随后利用异种移植和尾静脉注射肺转移模型来探究DUB-IN-3在体内肝癌中的作用,结果表明DUB-IN-3显著抑制了体内肿瘤的生长和转移(图2G-L)。这些数据表明DUB-IN-3可能通过靶向USP8作为一种新型有效的抗HCC药物。
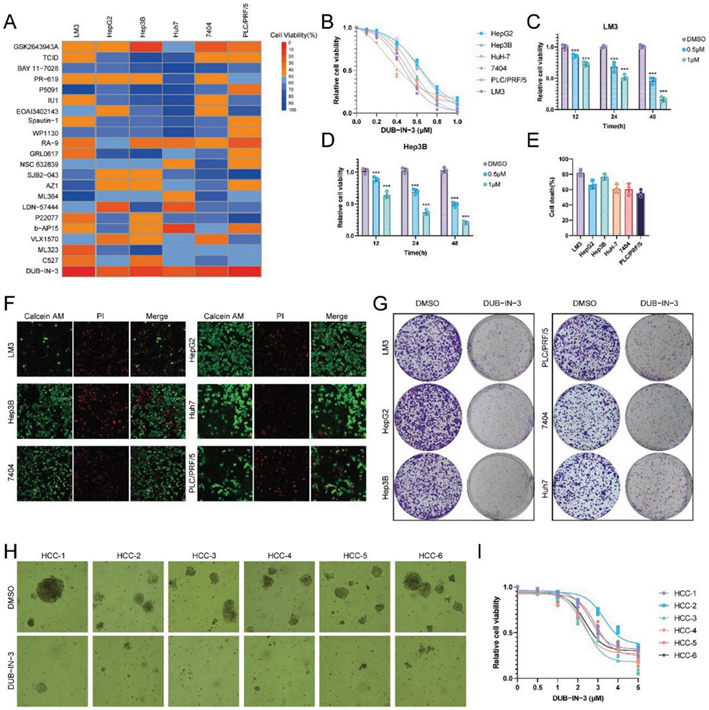
图1:DUB-IN-3是HCC的有效抑制剂
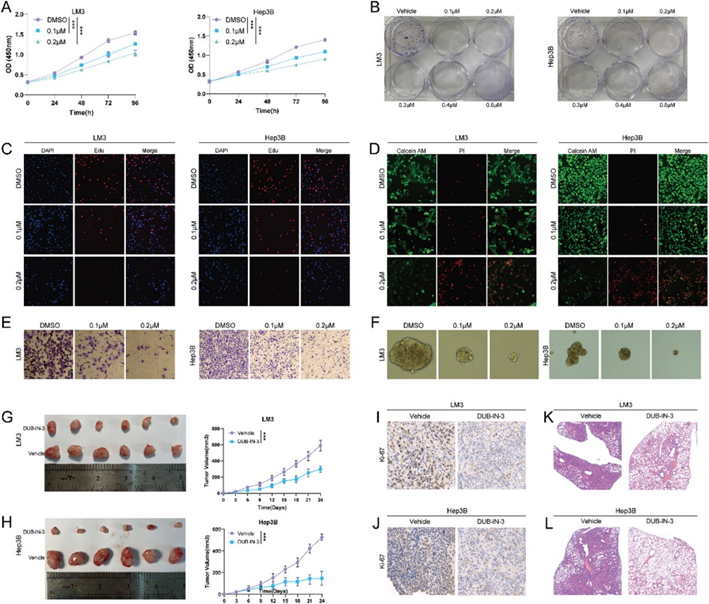
图2:DUB-IN-3在体内外均可抑制HCC进展
2. 靶向USP8抑制谷胱甘肽代谢并诱导HCC铁死亡
为了探索上述结果的潜在机制,作者进行了气相色谱法和基于TOF-质谱(GC/TOF-MS)的代谢组学分析。热图分析表明,DUB-IN-3处理引起了代谢产物的显著差异(图3A)。对代谢产物的途径进行富集发现,DUB-IN-3处理的细胞中谷胱甘肽(GSH)代谢途径、半胱氨酸和甲硫氨酸代谢途径以及铁死亡途径受到显著影响(图3B)。GSH是生物体内最重要的抗氧化剂之一,GSH代谢在肿瘤进展中起着重要作用,GSH水平升高与转移增加有关。有研究报告胱氨酸/谷氨酸反向转运蛋白Xc-/GSH/ GPX4轴是参与调节铁死亡的关键途径,这是一种新定义的程序性细胞死亡形式(图3C)。
靶向GSH的合成和运用被认为是肿瘤治疗的一种潜在手段。因此作者对GSH代谢途径进行研究。GSH代谢的关键中间产物如胱氨酸和谷胱甘肽在DIB-IN-3处理的细胞中显著降低(图3D,E)。DIB-IN-3处理后,细胞中亚铁水平上调(图3F)。GSH耗竭导致细胞积累更多的脂质ROS(图3G)。通过检测RSL3诱导的铁死亡进一步评估DIB-IN-3在铁死亡中的作用。DUB-IN-3降低了RSL3治疗下的细胞活力,可以通过Ferrostin-1挽救,表明该机制在细胞铁死亡中具有特异性(图3H,I)。敲除USP8降低了胱氨酸摄取和GSH水平,增加了亚铁铁水平和脂质ROS(图3J-M)。USP8耗尽的细胞对RSL3处理更敏感(图3N,O)。综上所述,药理抑制或敲除USP8可能通过抑制HCC细胞从细胞外环境吸收胱氨酸并赋予铁死亡来抑制GSH生物合成。
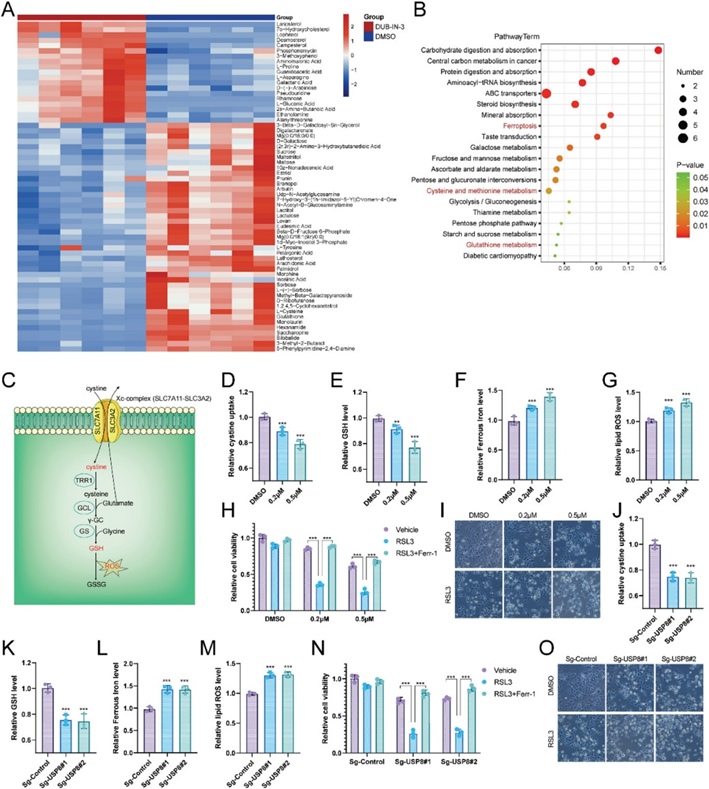
图3: 靶向USP8抑制GSH代谢并导致HCC发生铁死亡
3. USP8通过去泛素化调节OGT稳定性
为了找到可能由USP8调节的蛋白质,作者在Flag-USP8表达和免疫沉淀后进行了基于免疫沉淀的质谱分析(IP-MS),发现了USP8免疫沉淀的OGT蛋白(图4A)。进一步的Co-IP实验表明内源性USP8可以与内源性OGT互作(图4B)。GST-pulldown实验表明USP8在体外与OGT互作(图4C)。接下来作者通过免疫荧光证明了USP8和OGT在细胞内共定位。此外,蛋白的结构域和缺失突变体的Co-IP结果显示OGT的GT结构域与USP8的USP结构域存在物理相互作用(图4E-H)。
由于USP8是泛素特异性加工蛋白酶家族的成员,作者假设USP8可能通过泛素-蛋白酶体通路调节OGT周转。USP8的消耗显著降低OGT表达,却不影响其mRNA丰度(图5A),并且通过添加蛋白酶体抑制剂MG132或过表达USP8-WT可以逆转OGT下降趋势,而USP8催化失活突变体(USP8-C786A)不能逆转OGT下降趋势(图5B,C)。因此作者认为USP8影响了OGT的稳定性,为了证明这一结论,蛋白合成抑制剂放线菌酮(CHX)处理LM3细胞,USP8敲低后OGT半衰期缩短(图5D)。OGT稳定性在过表达USP8-WT时增加,在USP8-C786A时没有增加(图5E)。这些结果表明USP8通过泛素-蛋白酶体途径稳定细胞中的OGT。进一步研究OGT是否是USP8的底物。如图5F所示,USP8的耗竭显著提高泛素化OGT的水平;USP8-WT的表达显著降低泛素化OGT(图5G)。作者还对突变泛素(K6、K11、K27、K29、K33、K48和K63)进行泛素化测定,结果表明USP8可以有效地从OGT蛋白中去除K48连接的泛素链(图5H)。为了确定被USP8去泛素化的OGT蛋白的特定位点,作者突变了OGT的赖氨酸残基(图5I),泛素化测定表明K117是关键位点(图5J)。综上所述,这些结果表明USP8是OGT的调节因子,是预后标志物。
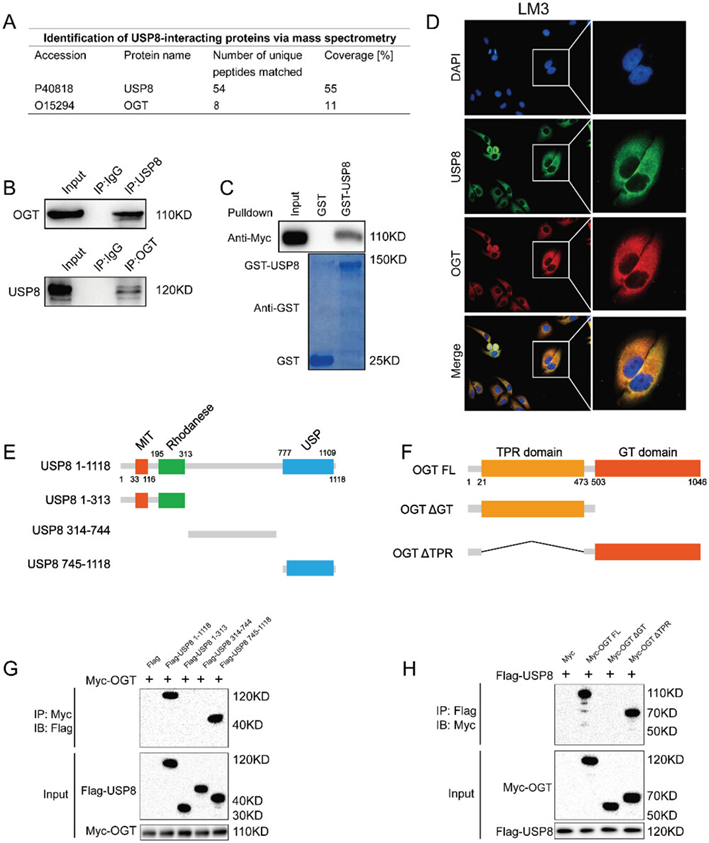
图4: USP8与OGT相关
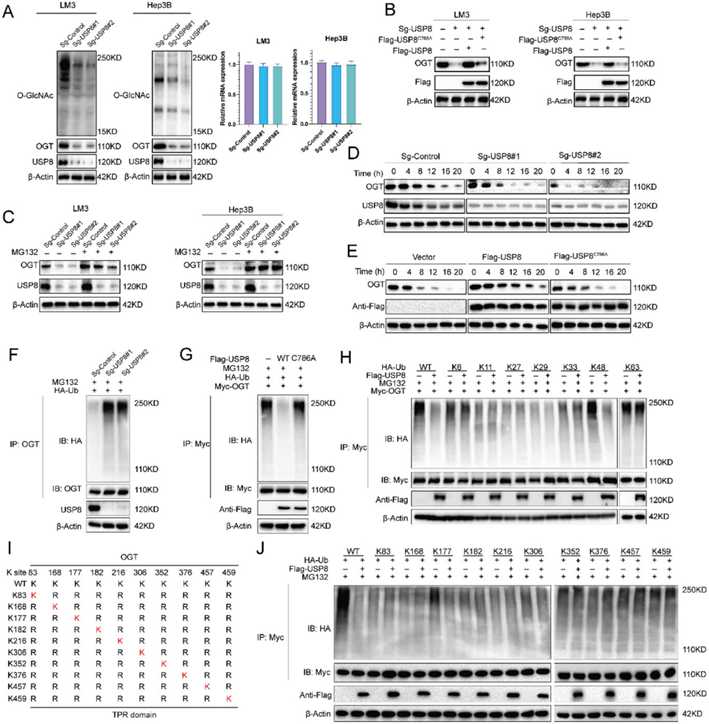
图5: USP8稳定去泛素化酶OGT
4. USP8的磷酸化是与OGT相互作用所必需的
作者发现USP8在Ser716残基处出现磷酸化,而且STE20样激酶(SLK)与Flag-USP8共纯化。在LM3细胞中进行Co-IP验证SLK和USP8之间发生互作(图6A)。敲低SLK显著降低了USP8的S716磷酸化(图6B,C)。接下来,作者探究了SLK介导的USP8磷酸化是否影响其与OGT的关联。敲低SLK后USP8和OGT之间的关联显著降低(图6D)。USP8 S716无义磷酸化突变体)(USP8 S716A)与OGT之间的关联下降,USP8 S716类似磷酸化(USP8 S716D)与OGT之间的关联升高(图6E)。随后,作者表达USP8- WT或USP8 S716D的LM3细胞中敲低SLK发现只有USP8-WT与OGT结合的能力降低(图6F)。作者还观察到USP8- WT和USP8 S716D增加了OGT蛋白表达水平(图6G)。
为了研究USP8磷酸化在HCC细胞中的作用,作者在HCC细胞中稳定表达USP8 WT和S716A,与对照相比,USP8 WT促进了HCC细胞的增殖、侵袭和干性,增强了HCC细胞的胱氨酸摄取和GSH水平,降低了脂质ROS水平和细胞对RSL3治疗的敏感性,而USP8 S716A突变体失去了这些能力(图6H-P)。USP8 WT,但不是C786A和S716A突变体,(图6M-P)。这些结果表明USP8与OGT相互作用时需要磷酸化激活。
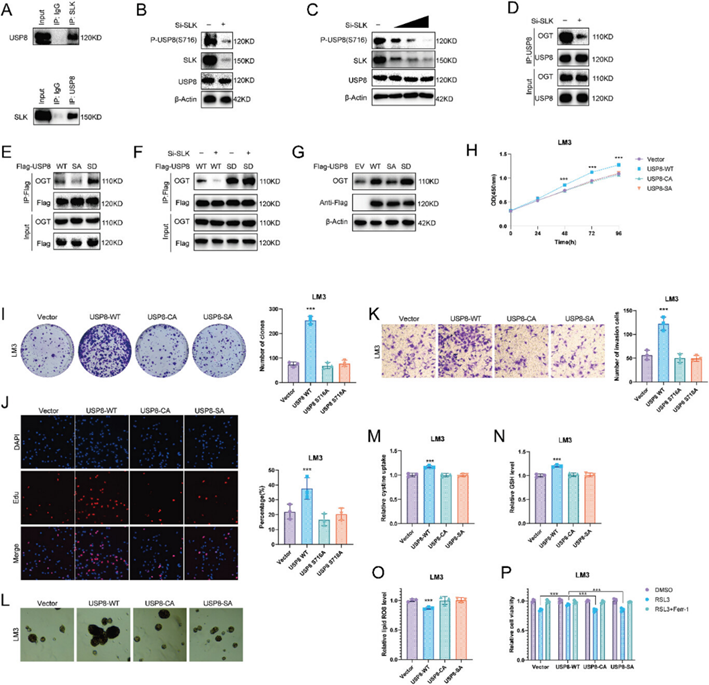
图6: SLK磷酸化USP8
5. HCC细胞中OGT O-GlcNAa糖基化SLC7A11 Ser26
作者已经证明USP8去泛素化OGT并影响HCC细胞的胱氨酸摄取。由于胱氨酸是由胱氨酸/谷氨酸反向转运蛋白(System Xc-)从卵泡外环境输入的,故作者研究该反向转运蛋白的功能亚基——SLC7A11是否受USP8-OGT轴的调节。Co-IP表明OGT和SLC7A11在生理条件下存在关联(图7A);SLC7A11发生了O-GlcNAa糖基化,OGT的消耗可降低SLC7A11的O-GlcNAa糖基化(图7B)。TMG(OGA抑制剂)处理细胞发现整体O-GlcNAa糖基化的升高增加了SLC7A11的O-GlcNAa糖基化,OSMI-1(OGT抑制剂)处理的细胞显示出相反的现象(图7C)。SLC7A11的O-GlcNAa糖基化水平在DAB-IN-3处理或敲除USP8后下降(图7D,E)。此外,OGT的过表达可以挽救USP8-KO细胞中SLC7A11的O-GlcNAa糖基化(图7F)。这些结果表明USP8通过OGT调节SLC7A11的O-GlcNAa糖基化水平。
为了确定SLC7A11上发生O-GlcNAa糖基化的位点,作者使用在线生物信息学工具(https://services.healthtech.dtu.dk/)进行预测,确定了4个残基(S8,T9,S26和T218)。为了确定SLC7A11中糖基化的发生位点,将四个残基均突变为丙氨酸,发现S26的突变降低了O-GlcNAa糖基化水平,表明S26是主要的糖基化位点(图7G,H)。为了进一步了解O-GlcNAa糖基化对SLC7A11活性的影响,在HCC细胞中敲除了SLC7A11(SLC7A11-KO),转染SLC7A11-WT和SLC7A11-S26A。结果发现敲除SLC7A11抑制HCC细胞的增殖、侵袭和干性,SLC7A11-WT的过表达恢复了SLC7A11敲除诱导的效果,SLC7A11-S26A不能恢复(图7I-N)。类似的,SLC7A11-WT增强了HCC细胞的胱氨酸摄取和GSH水平,同时降低了脂质ROS水平和细胞对RSL3治疗的敏感性(图7O-R)。综上所述,SLC7A11在HCC细胞中被OGT进行O-GlcNAa糖基化,SLC7A11的O-GlcNAa糖基化对其胱氨酸摄取活性至关重要。
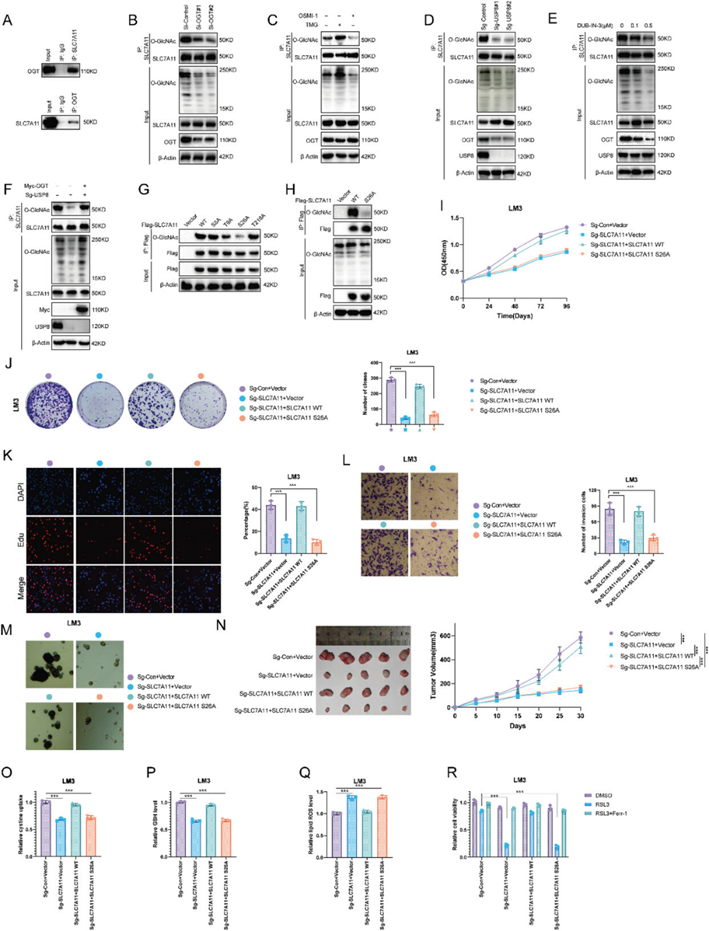
图7:HCC细胞中OGT通过O-GlcNAc糖基化修饰SLC7A11 Ser26位点
结论
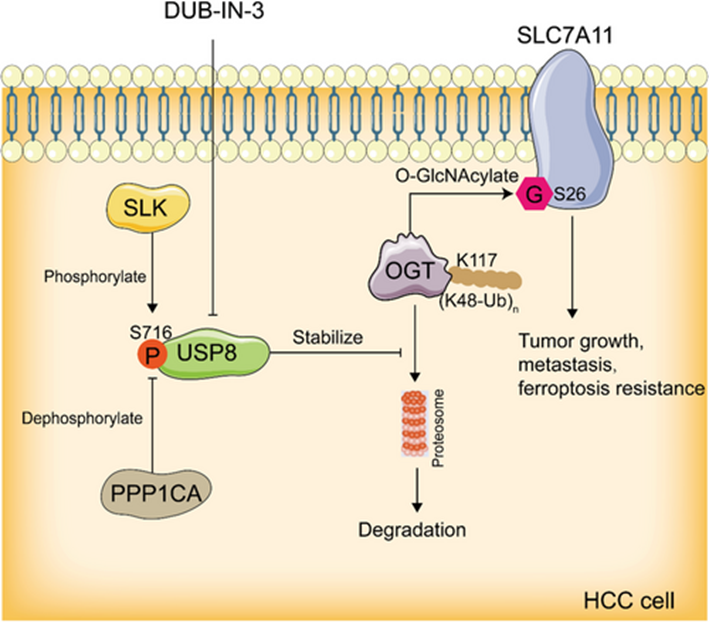
总之,本研究证明了靶向USP8降低了OGT的稳定性,抑制了肝癌的进展,并诱导了肝癌的铁死亡。从机制上讲,USP8通过去除K48连接的泛素链使OGT去泛素化和稳定增加,然后SLC7A11在S26位点发生O-GlcNAc糖基化。SLC7A11的O-GlcNAc糖基化是其从细胞外环境中吸收胱氨酸的活性所必需的。此外,USP8在S716的磷酸化是与OGT相互作用所必需的。我们的发现为USP8在肝癌中调节谷胱甘肽代谢和铁死亡的作用提供了新的见解(图8),并表明靶向USP8是一种有希望的肝癌治疗策略。
实验方法
代谢组学分析,蛋白质稳定性分析,去泛素化实验,蛋白质印迹分析,脂质ROS分析,GSH检测,细胞培养,质粒和USP8敲除细胞系的生成,细胞染色,细胞活力测定,球体形成分析,小鼠实验,体内肿瘤发生试验
参考文献
Tang Jianing, Long Guo, Hu Kuan et al. Targeting USP8 Inhibits O-GlcNAcylation of SLC7A11 to Promote Ferroptosis of Hepatocellular Carcinoma via Stabilization of OGT. [J]. Adv Sci (Weinh), 2023, 10: e2302953.