






在恶性横纹肌样肿瘤中, SMARCB1缺失激活了患者特异性远端致癌增强子
恶性横纹肌样瘤(MRT)是一种高度恶性且通常致命的儿童癌症。 MRT 在基因上由 SMARCB1 的双等位基因失活突变定义,SMARCB1 是 BRG1/BRM 相关因子 (BAF) 染色质重塑复合体的成员。 BAF 复合体成员的突变在人类癌症中很常见,但在许多情况下,人们对它们对肿瘤发生的贡献仍知之甚少。在这里,我们研究了 MRT 背景下 SMARCB1 丢失导致的监管环境脱轨。我们对源自患者的 MRT 类器官的多组学方法揭示了 SMARCB1 重组后监管格局的巨大重塑。染色体构象捕获实验随后揭示了远端增强子区域与 MYC 癌基因启动子的患者特异性环。联合单细胞 RNA-seq 和 ATAC-seq 显示,这种 MYC 增强子利用的肿瘤间异质性也存在于患者 MRT 组织中。我们发现 SMARCB1 的缺失会激活 MRT 肿瘤发生背后的患者特异性表观遗传重编程。
该研究于2023年12月发表在《Nature communications》,IF:16.6。
结果:
1、MRT PDOs 中的染色质动力学分析揭示了 SMARCB1 依赖性增强子调节
我们先前证明了SMARCB1的丧失是恶性转化所必需但不足够的。因此,我们假设肿瘤形成需要额外的表观遗传学驱动因子。将MRT的主要遗传驱动事件SMARCB1丧失重新引入,将MRT细胞恢复到正常状态,表明促使恶性的表观遗传变化是可以克服的(图1A)。为了找到这些促使肿瘤发展的调控性变化,我们对一种MRT PDO模型(命名为P103)进行了慢病毒转导,分别使用荧光素酶表达(对照)或SMARCB1表达(SMARCB1+)质粒,通过转座酶可及染色质测序(ATAC-seq)以及染色质免疫共沉淀测序(ChIP-seq)或使用核酸酶测序进行靶点切割和释放测序(CUT&RUN)来测量染色质可及性(图1A、B)。在SMARCB1重建后,我们发现了7,941个新形成的开放染色质区域(OCRs),这些区域富集了来自不同家族的转录因子结合位点,如SMARCC1/2和AP-1(图1C)。SMARCB1 ChIP-seq显示这些OCR被SMARCB1(cBAF和PBAF)和SS18(ncBAF和cBAF)结合,表明在重建后这些区域存在cBAF的结合。当我们使用GREAT对这些OCR进行功能注释时,发现多个类别富集,主要与分化和发育过程相关。在失去的1,211个OCR中,观察到了ncBAF复合物成员BRD9和SS18的结合明显减少。我们发现这些区域中唯一富集的基序是隔离蛋白CTCF的基序,与之前在人类胚胎干细胞和小鼠胚胎成纤维细胞中的报告一致
CTCF的ChIP-seq结果显示,在SMARCB1重建后,CTCF在SMARCB1+细胞中失去的OCR上的结合减少(图1C)。在基因组中,CTCF的结合经常与环形粘连复合物的结合重叠。粘连复合物亚单位RAD21的ChIP-seq证实,除了CTCF结合减少外,SMARCB1+细胞中失去的OCR显示了粘连复合物的结合减少(图1C)。另一方面,在SMARCB1+ PDO中增益的OCR显示了RAD21的占有(图1C),但没有CTCF的结合。为了确定SMARCB1重建后增强子景观的变化,我们进行了H3K27ac活性增强子标记的ChIP-seq。如预期,失去的OCR显示H3K27ac水平下降,而增加的可及性位点则显示H3K27ac水平显著增加(图1C)。活性增强子标记的增加与粘连复合物的结合增加相一致(图1C),这与一部分粘连复合物分子结合到(超级)增强子区域以及CTCF结合位点一致。这些结果表明,经典BAF复合物在抑制CTCF结合到染色质方面起着重要作用,并且SMARCB1的丧失导致CTCF结合到染色质的可及性增加,以及ncBAF复合物结合到肿瘤特异OCR。
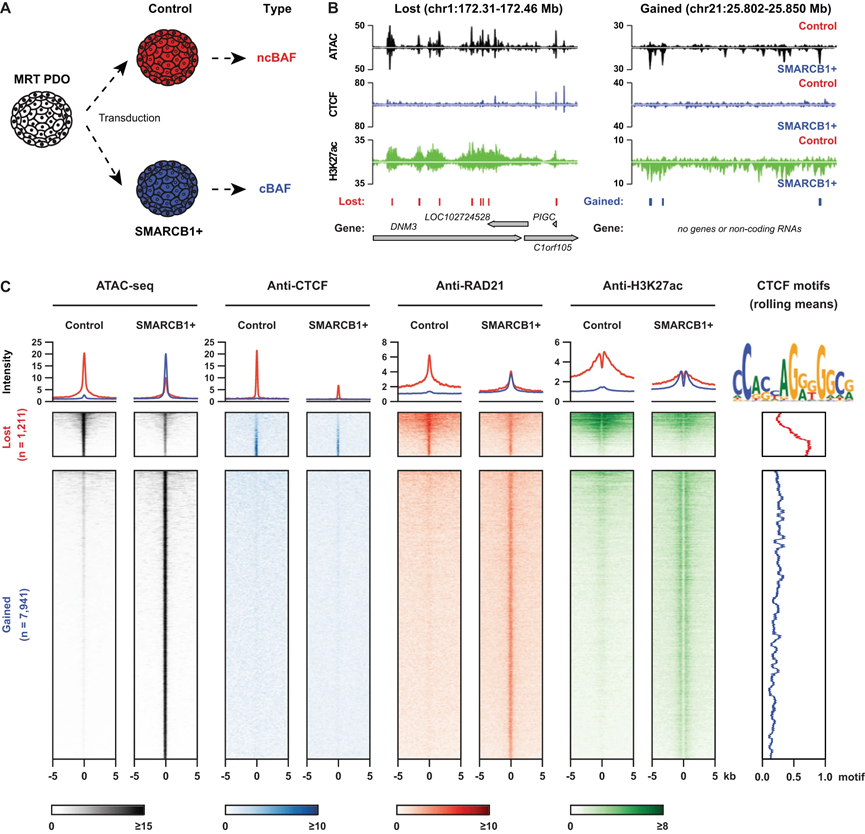
图1| SMARCB1 重建重塑了 MRT PDO 模型中开放的染色质景观。
2、SMARCB1 重建重组了 MYC 癌基因周围的染色质景观
观察到在SMARCB1重建后CTCF和RAD21结合景观的变化使我们假设,这些细胞中可能影响了长距离基因调控。为了确定基因组的组织(3D基因组)是否发生了任何依赖于SMARCB1的变化,我们在对照和SMARCB1+ P103 MRT PDOs中进行了高分辨率原位HiC实验。尽管CTCF对染色质的结合存在差异,但我们发现染色质结构基本上没有受到影响。3D基因组特征,如拓扑相关域(TADs)和A/B区,SMARCB1重建后显示出有限的变化。然而,我们分别鉴定了控制和SMARCB1+ PDO的131个和1,164个特异loop,其最小接触频率变化为1.5倍(图2C)。在SMARCB1重建后,对最显著失去的基因座进行排名,我们发现MYC癌基因的启动子与一个大约1.1 Mb的远程区域之间存在相互作用(图2A,C)。这个loop是SMARCB1重建后第六个最减少的相互作用,也是涉及原癌基因的排名最高的相互作用。这个MYC启动子的远程区域具有高H3K27ac水平,表明这是一个超级增强子(图2A,B)。
在MRT的背景下,MYC癌基因尤为引人关注,因为我们和其他研究先前证明,MRT是通过SMARCB1依赖的MYC基因表达特征来定义的。类似地,我们观察到在SMARCB1重建后MYC mRNA和蛋白水平明显减少,以及细胞周期相关基因的失调,表明存在细胞周期停滞(图2D)。此外,通过shRNA诱导的MYC敲除在MRT PDOs中导致细胞增殖显著减少(图2E),表明MYC对MRT的生长是必需的。除了MYC表达的减少外,我们观察到在SMARCB1重建后,超级增强子以及MYC启动子的染色质可及性显著减弱(图2A,B)。为了探讨SMARCB1重建是否影响ncBAF复合物的结合,我们对BRD9(ncBAF)和SS18(cBAF和ncBAF)进行了CUT&RUN。我们发现在SMARCB1重建后,BRD9和SS18在MYC启动子以及1.1 Mb远程区域的结合显著减少(图2B)。这些结果表明,MYC表达至少在很大程度上取决于ncBAF复合物的结合。SMARCB1的重建减少了ncBAF复合物在MYC位点的结合,从而可能减少了MYC启动子与远程超级增强子的相互作用。
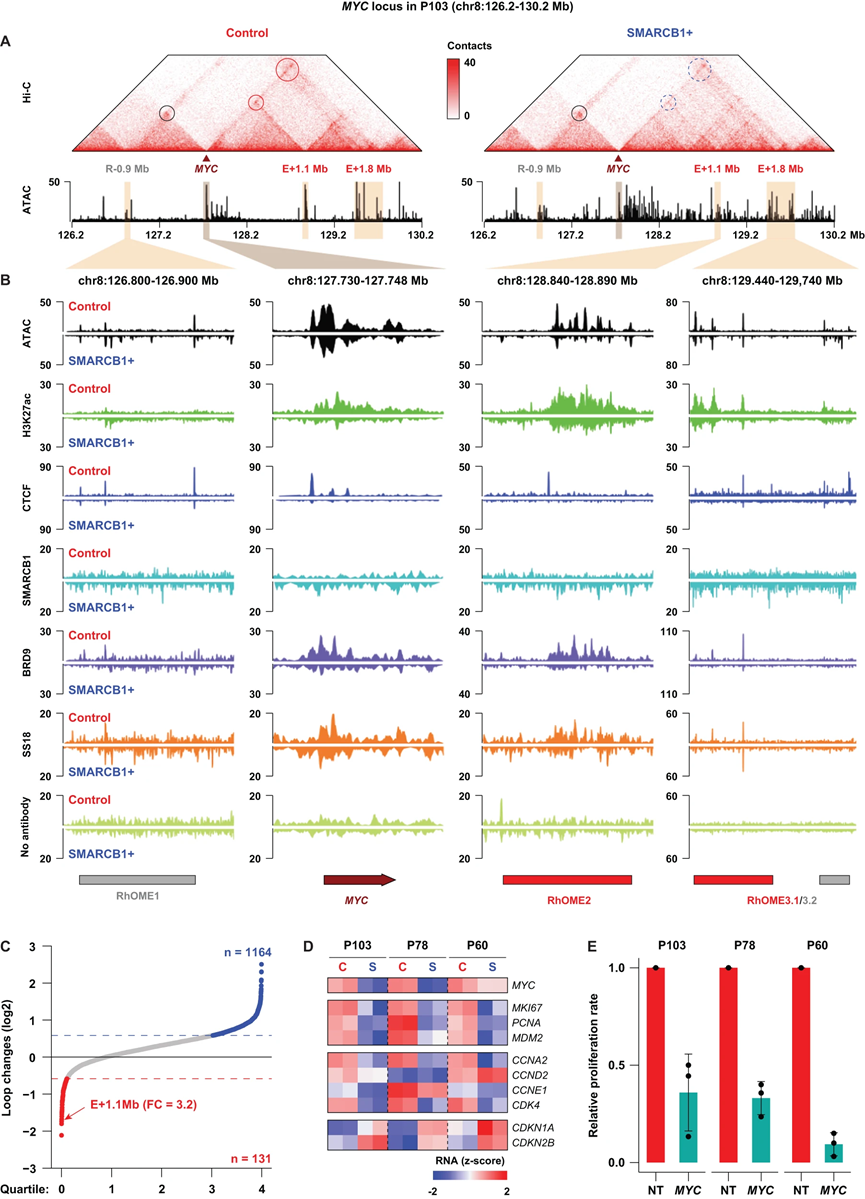
图2| SMARCB1 重建影响 MRT PDO 中 MYC 癌基因的远端增强子
3、患者特异性超级增强子与 MRT 中的 MYC 相互作用
我们随后假设MYC远程增强子环路可能是推动MRT致癌的一种普遍机制。为此,我们在另外两个MRT PDOs(P78和P60)中进行了SMARCB1的重建。RNA-seq分析显示这两个MRT模型中MYC表达减少,以及细胞周期基因的失调,证实了细胞增殖停滞的诱导(图2D、E)。接下来,我们使用MYC启动子作为视点,对这两个额外的PDO进行了4C-seq,这是一种定向到特定基因组位点的染色体构象捕获方法。与P103类似,P78在SMARCB1重建后显示了MYC启动子与相同的约1.1 Mb远程基因组区域的减少接触频率(图3A)。值得注意的是,第三个PDO模型(P60)的4C-seq分析显示MYC启动子与我们先前确定的基因组区域没有相互作用。相反,在SMARCB1重建后,4C-seq揭示了另外两个染色质环路的减弱(图3A)。
为了绘制PDO模型在存在和不存在SMARCB1时的调控景观,我们在P78和P60上进行了ATAC-seq。当我们将ATAC-seq数据与4C-seq数据叠加时,发现与MYC启动子相互作用的区域是高度可访问的。这种可访问性与患者特异的MYC启动子相互作用景观相关(图3B)。在SMARCB1重建后,相互作用的丧失与这些位点的可访问性丧失相一致(图3B)。在P78中的可访问染色质区域类似于我们在P103中确定的超级增强子。总体而言,我们将这些可能的超级增强子区域称为Rhabdoid Oncogenic MYC Enhancer(RhOME),并根据它们在基因组中的位置进行编号。值得注意的是,与RhOME2相反,RhOME1(PCAT1)和RhOME3(CCDC26)先前在其他肿瘤实体中已被确定为MYC的调控区域。因此,我们的Hi-C和4C实验揭示,在不同的MRT PDOs中,与MYC启动子相互作用的不同远程增强子是活跃的(RhOME1-3)。RhOMEs的肿瘤间特异性进一步突显了增强子RNA(eRNAs)的表达,这是活跃的超级增强子所特有的,特异地在这些增强子在MRT PDOs中是活跃的并与MYC启动子相互作用的情况下(图3C)。这些结果表明,SMARCB1的丧失导致了一种表观状态,其特征是形成长程启动子-增强子环路,这些环路对于PDO模型是特异的,但在来自不同供体的MRT PDOs之间是异质的。
因为MRT PDOs在很大程度上保留了它们衍生自的组织的(表观)遗传特征,我们利用这些模型进一步探索患者特异的表观遗传程序,使用ATAC-seq(图4A)。为了对在P103中丧失的OCR进行分层,我们执行了K均值聚类,包括来自三个PDOs的ATAC-seq数据和P103的ChIP-seq数据(图4A)。因此,我们可以将失去的OCR分类为(K1)非增强子CTCF结合位点、(K2)弱增强子和(K3)独立于CTCF的超级增强子(图4A)。与超级增强子在细胞身份控制中的功能一致,我们鉴定了SOX蛋白结合位点,包括SOX2、SOX9和SOX17,以及与K3簇特异性开放染色质位点、RhOME2和3.1 OCR相关的许多发育过程的功能注释。此外,K3簇显示了ncBAF复合物的最高染色质占有率,由BRD9 CUT&RUN测量,其在恢复SMARCB1表达后显著减少。而K1和K2簇在三个PDO模型中显示了相似的可访问性模式,K3簇的可访问性丧失是特定于P103细胞系的(图4A)。我们在P103中的Hi-C分析显示,这些K3簇特异性超级增强子形成了绝缘边界,这在重建SMARCB1表达后被有效地废除(图4B、C)。因此,在MRT PDOs中的SMARCB1重建与基因组拓扑、BAF复合物占有率和增强子活性方面发生了戏剧性但局部、患者特异性的变化。
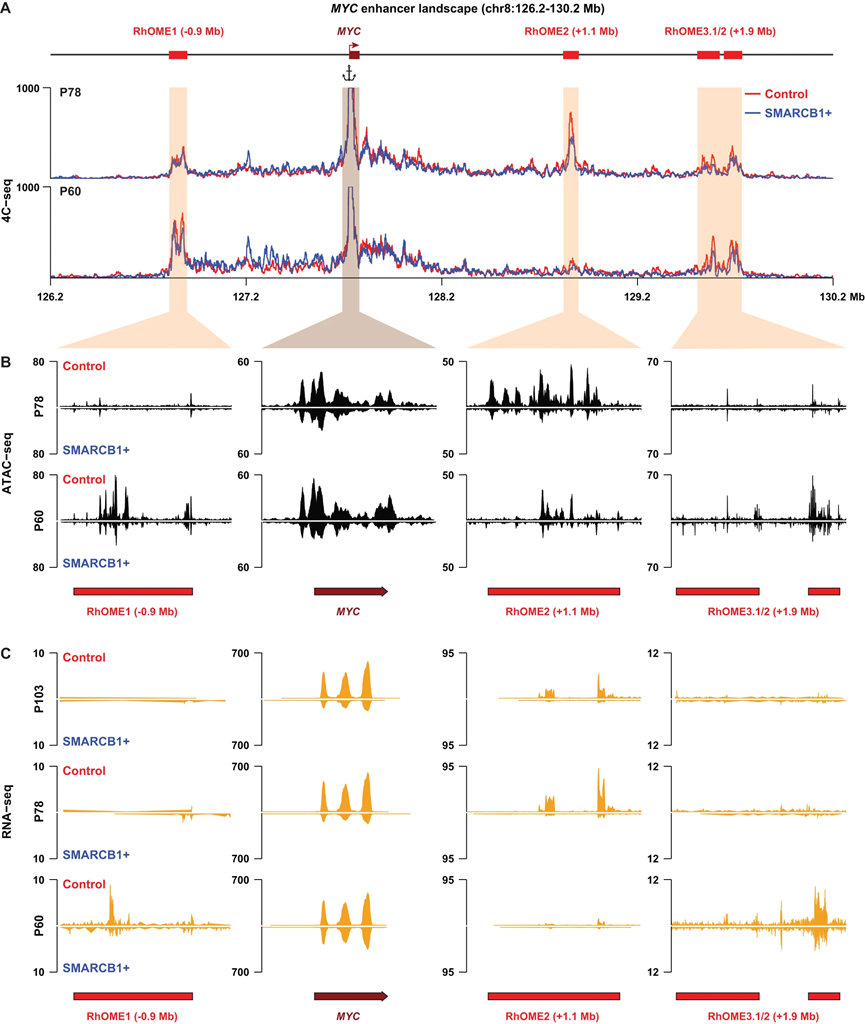
图3|不同 MRT PDO 中患者特异性 MYC 增强子组的鉴定。
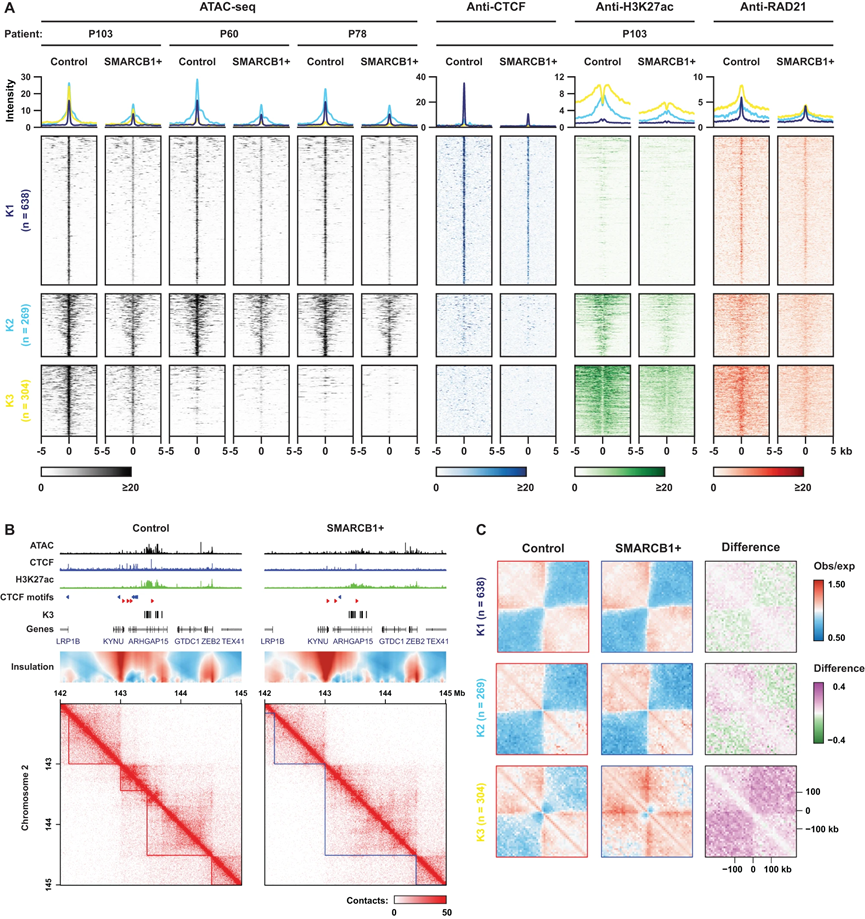
图4|患者特异性超级增强子在 SMARCB1null 肿瘤 PDO 中形成独立于 CTCF 的 TAD 边界。
4、MYC 增强子可塑性反映在患者 MRT 中
到目前为止,我们的数据引发了一个有趣的可能性,即在增强子利用的水平上存在推动MYC癌基因表达的肿瘤间异质性。为了将我们的体外发现推广到患者组织,我们首先检查了来自患者MRT组织的公开可获得的H3K27ac ChIP-seq数据。如预期,MYC启动子在大多数患者样本中都标有高水平的H3K27ac,表明活跃的转录(图5A)。此外,RhOME1-3处广泛富集H3K27ac,显示了在MRT PDOs中鉴定的肿瘤超级增强子景观在MRT组织中的保留。在STJ0090和STJ0537中未能检测到RhOME1-3的清晰峰值,而在MYC启动子处检测到弱的H3K27ac信号。值得注意的是,与我们的PDO数据一致,不同患者样本中在不同的RhOME位点检测到H3K27ac峰的存在呈异质性分布(图5A)。
为了评估是否在肿瘤内存在差异的增强子利用,即肿瘤内异质性,我们利用单细胞Multiome(10x Genomics)对七个MRT患者组织样本进行了联合单细胞RNA-seq和ATAC-seq(图5B)。经过过滤,留下14,799个细胞用于进一步的分析,每个肿瘤中的中位数为2040个细胞(图5C)。通过细胞类型标记基因41和SMARCB1表达将正常和肿瘤细胞进行了分配。均匀流形逼近和投影(UMAP)空间显示,肿瘤细胞按患者进行了聚类,而非恶性细胞按细胞类型进行了聚类(图5C)。MYC表达以及MYC启动子的OCR在所有MRT和一些正常细胞聚类中都有发现(图5D)。然而,尽管在正常细胞中检测到可察觉的MYC启动子信号,但几乎在RhOME1-3中没有检测到可访问的染色质,这表明在至少这些样本中,这些超级增强子是肿瘤特异性的。至关重要的是,我们观察到在不同的患者MRTs中,RhOME1-3的OCR组合是不同的(图5D、E)。更具体地说,尽管P156和P168仅由RhOME2处的OCR定义,但P041和P052主要显示RhOME1和RhOME3处的OCR,并且在P166中仅在RhOME3处检测到OCR。在P116和P138中在任何RhOME中都没有发现OCR,这表明在这些肿瘤中通过RhOME1-3以外的调控元件发生了超生理激活MYC。总体而言,这些结果表明,在MRT中,存在在超级增强子活性水平上推动MYC癌基因表达的肿瘤间异质性。
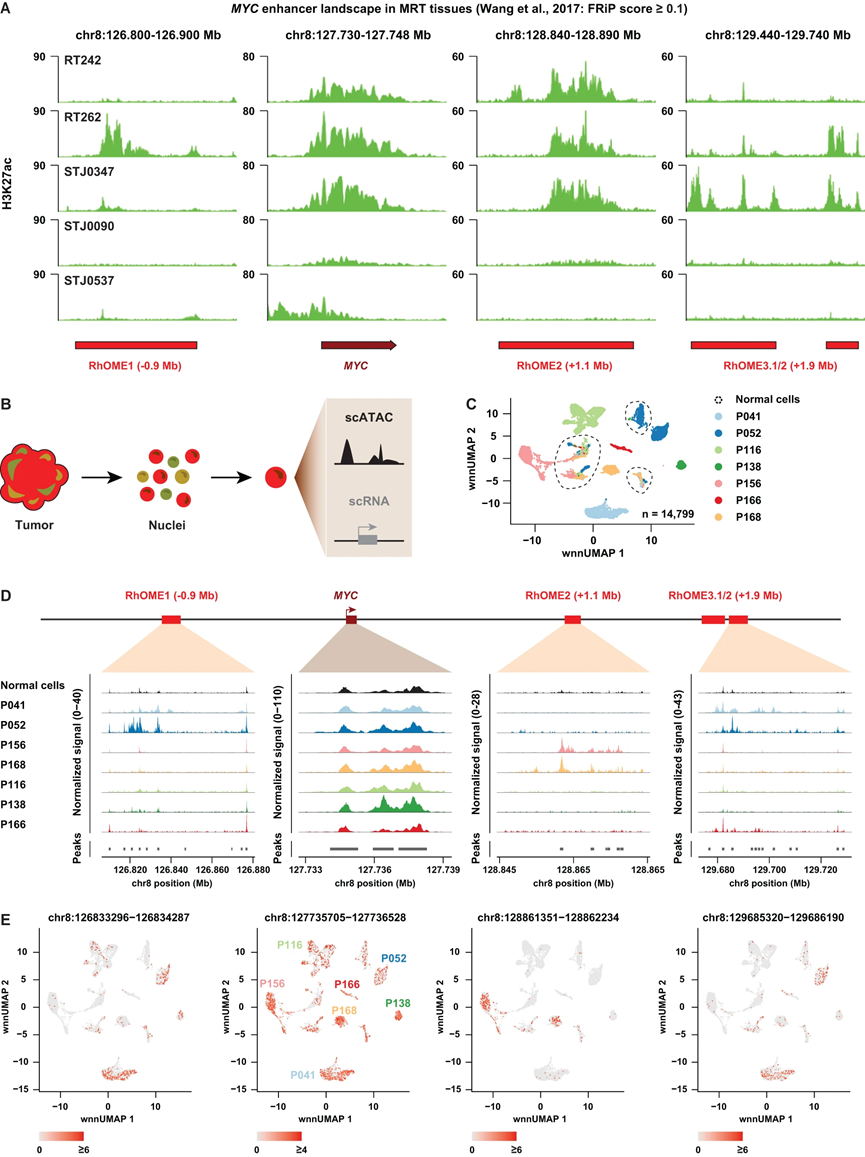
图5 | MRT 组织分析揭示了 MYC 增强子景观的肿瘤间和肿瘤内异质性
5、ncBAF 抑制诱导类似于 SMARCB1 重建的基因表达特征
最近的研究表明,MRT可能由ncBAF复合物在MRT细胞中异常定位所驱动。因此,抑制ncBAF亚单位BRD9被证明是MRT中的一个潜在治疗易感性。因此,我们着手研究ncBAF抑制对MRT PDOs的影响(图6A)。为此,我们使用BRD9的药物抑制剂(I-BRD9)处理MRT PDOs。我们观察到,在形态上,MRT细胞呈现出与SMARCB1重建相似的分化表型(图6A),并且细胞生长显著受到抑制。定量RT-PCR(RT-qPCR)证实,BRD9抑制导致MYC mRNA水平显著下降(图6B)。我们进行了RNA-seq以进一步测量ncBAF抑制后的转录组变化,并发现I-BRD9处理后引起的基因表达变化与SMARCB1重建后的变化之间存在显著的关联(图6C)。为了在两个基因集之间统计确认差异,我们对这两个集合进行了Fisher确切性检验(图6D),确认了强烈的相似性。与增殖下降一致,我们观察到细胞周期相关基因集以及MYC靶基因的下调(图6E)。在上调的基因集中,我们发现了与分化相关的基因的富集(图6E),与用BRD9抑制剂处理的细胞的形态表型一致(图6A)。BRD9和SS18的CUT&RUN证实了在MYC启动子以及RhOME2和3位点的ncBAF复合物结合的减少(图6F)。更一般地,在I-BRD9处理后观察到BRD9和SS18的基因组范围内结合的丧失,这并不是由于处理引起的BRD9和SS18蛋白表达的降低引起的。因此,证实了治疗诱导的ncBAF复合物结合的丧失。因此,在MRT中抑制ncBAF复合物,消除了所有三个BAF复合物,呈现出重建SMARCB1表达的效应的表型。
总的来说,我们的全面研究表明,由于SMARCB1丧失导致超级增强子活性的偏离支撑了MRT的肿瘤发生,并为揭示BAF复合物突变在各种癌症中对肿瘤发生的贡献提供了蓝图。
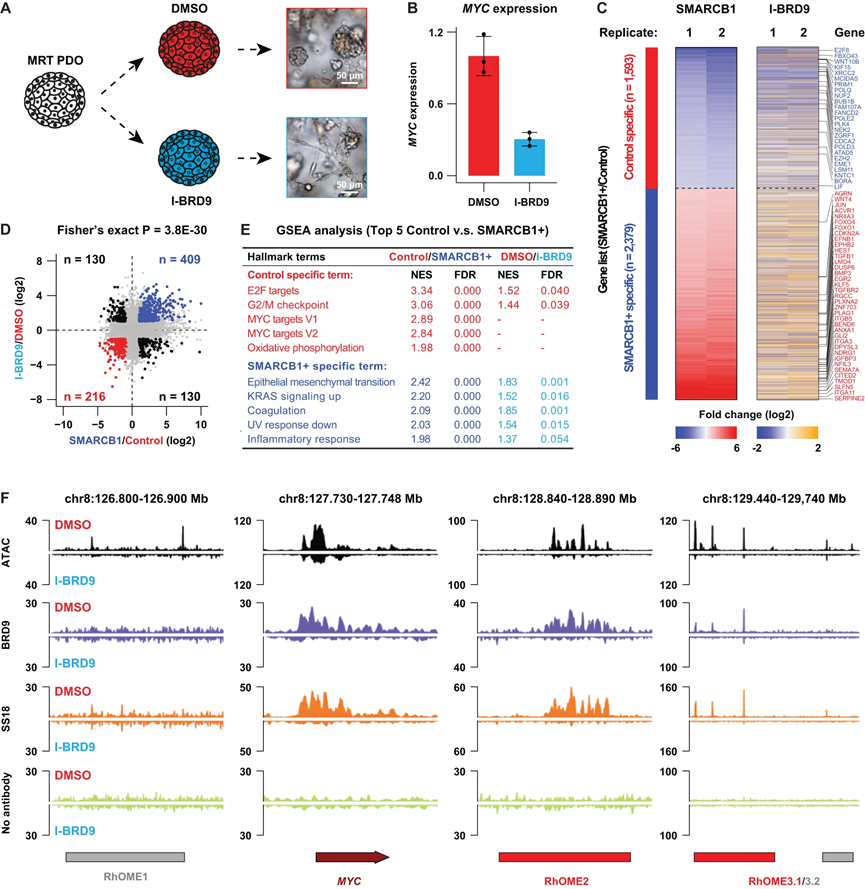
图 6 |通过使用药物抑制剂靶向名为 BRD9 的 ncBAF 亚基来治疗 MRT 患者
实验方法:
慢病毒转导、定量RT-PCR、细胞活力测定、Western Blot、RNA-seq、ATAC-seq、ChIP-seq、CUT&RUN、Hi-C and 4C-seq、Motif分析、单细胞多组 ATAC + GEX。
参考文献:
Liu, N.Q., Paassen, I., Custers, L. et al. SMARCB1 loss activates patient-specific distal oncogenic enhancers in malignant rhabdoid tumors. Nat Commun 14, 7762 (2023). https://doi.org/10.1038/s41467-023-43498-3