






USP13通过改变肺棒状细胞谱系可塑性来驱动肺鳞状细胞癌
肺癌是最常见的癌症,也是全球癌症相关死亡的主要原因。非小细胞肺癌(NSCLC)和小细胞肺癌(SCLC)是两种最常见的肺癌。NSCLC占肺癌病例的85%以上,分为肺腺癌(LUAD,50%)、肺鳞状细胞癌(LUSC,30-40%)和大细胞癌。大多数LUSC患者被诊断时已处于晚期,靶向治疗有限。谱系可塑性即细胞从一种分化状态转变为不同身份的能力,与肿瘤内异质性、肿瘤亚型之间的组织学转变以及肺癌治疗耐药的潜在机制有关。细胞起源影响肿瘤的组织类型、恶性程度、谱系可塑性和肿瘤微环境。LUSC的起源细胞被假设为基底样干细胞、AT2细胞、细支气管肺泡干细胞和小气道的棒状细胞。细胞谱系特异性转录因子驱动了多种肺癌类型。人肺鳞癌由正常气道上皮发展而来,并通过增生、鳞状上皮化生、不典型增生和癌进展。然而并非所有的癌前病变都注定会进展为浸润性LUSC,尚不完全清楚哪些分子变化建立了每个阶段并驱动进展。超过90%的人类LUSC表现出染色体3q26拷贝数增加,这是LUSC的遗传标志。值得注意的是,这个3q26扩增子包含SOX2和许多其他潜在的驱动或修饰基因,这些基因可能与LUSC发生的生物学和治疗相关。USP13与人LUSC的癌基因PI3CA和SOX2一起位于染色体3q26扩增子上。USP13作为一种去泛素化酶,通过去泛素化过程调节多种底物蛋白的稳定性,从而促进或抑制肿瘤。该研究发表在《Molecular Cancer》,IF:37.3。
技术路线:

主要研究结果:
1. USP13在NSCLC中高扩增,且与不良预后相关
USP13基因拷贝数在人LUSC和其他几种癌症(包括卵巢癌、食管癌和头颈癌)中高度扩增(图1A)。癌症基因组图谱(TCGA)基因组学显示,91%(427例)的肺鳞状细胞癌和29%(145例)的肺腺癌中有USP13扩增(≥5拷贝)或获得(1~3拷贝)(图1B)。USP13与染色体3q26-28位点内的鳞状细胞谱系因子SOX2和TP63紧密定位,并在肺鳞状细胞癌和肺腺癌中共扩增(图1B)。在肺鳞状细胞癌和肺腺癌中,超过一半的USP13拷贝数增加/扩增与KRAS基因改变同时发生(图1B)。在肺鳞状细胞癌和肺腺癌中,USP13 mRNA水平随着基因拷贝数的增加而升高(图1C)。USP13 mRNA高表达的肺鳞癌患者无复发生存率较差。USP13高表达的肺腺癌患者总生存率降低(图1D)。在人类肺鳞癌和肺腺癌组织芯片中的免疫组织化学(IHC)分析显示,USP13蛋白在正常人类肺组织中几乎不表达,而肺鳞癌样本中USP13的表达水平是正常肺组织的16.7倍。USP13蛋白在所有级别的肺鳞状细胞癌患者中均升高(图1E)。在肺腺癌组织样本中,USP13的表达水平是正常癌旁组织的1.5倍(图1F)。
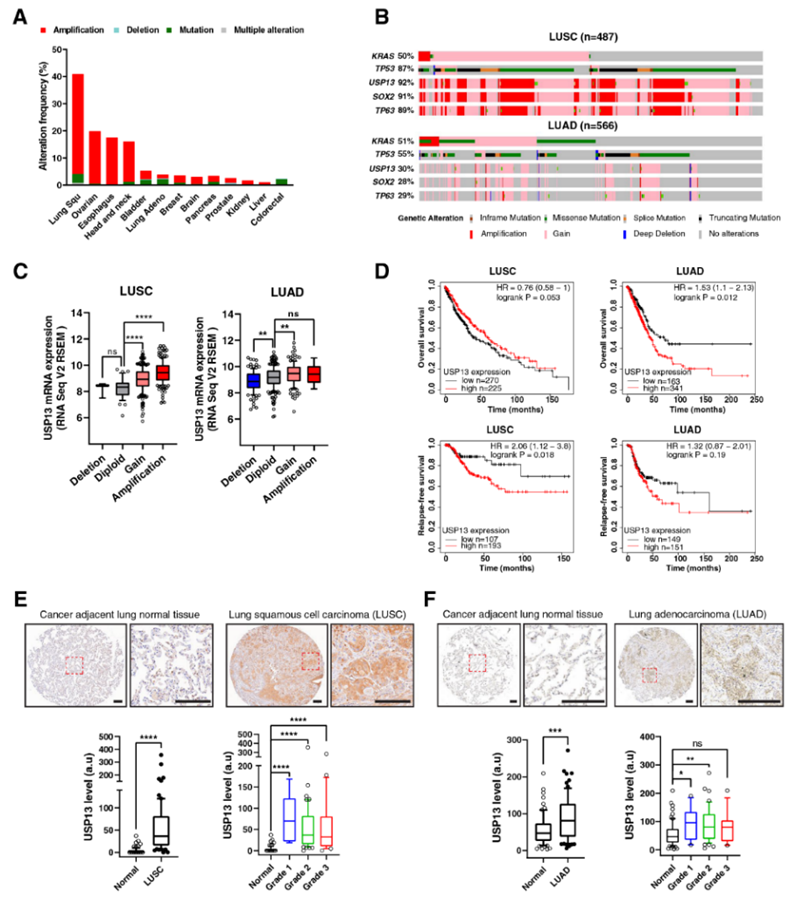
图1 USP13在NSCLC中高度扩增,并与不良生存相关
2. USP13驱动Kras/Trp53突变小鼠肺鳞状细胞癌的发展
为了研究3q26扩增子中的USP13在肺肿瘤发生中的作用,作者将USP13敲入(KI)小鼠模型(Usp13LSL/LSL,U)与KrasG12D/+(K)和Trp53flox/flox(P)小鼠杂交,产生KrasLSL-G12D/+;Trp53flox/flox;Usp13LSL/LSL(KPU)。通过在小鼠肺上皮内给予表达Cre重组酶的腺病毒(Ad5-CMV-Cre)诱导肺部肿瘤的发生,导致致癌性Kras活化、Trp53纯合缺失和Usp13过表达(图2A)。首先观察到KPU小鼠的生存期明显短于KrasG12D/+;Trp53flox/flox(KP)小鼠(图2B)。在Ad5-CMV-Cre病毒诱导后的10-14周,KP和KPU小鼠的所有肺叶均可检测到肿瘤。尽管产生的肿瘤结节较少,但KPU驱动的每个肿瘤的大小显著大于KP驱动的肿瘤(图2C,D)。
通过免疫组化分析进一步检查KP和KPU肿瘤。KP肿瘤表达LUAD标志物,包括NKX2-1和SPC(图2E)。KPU肿瘤强表达鳞状细胞标志物(SOX2和CK5),而腺癌标志物显著下调(图2E)。作者还通过免疫印迹分析证实了KP和KPU肿瘤裂解物中LUAD和LUSC标志物的差异表达。在KPU肿瘤溶解物中,NKX2-1和CK7信号被下调,而p63异构体的表达被改变,SOX2被上调(图2F)。根据免疫组织化学特征,对KP和KPU的每个肿瘤结节进行肿瘤亚型分型。所有KP肿瘤均被确定为腺癌(图2G)。两种主要的肺亚型LUAD和LUSC,在以鳞状组织为主的大尺寸肿瘤结节的KPU小鼠中观察到100%(图2G,H)。综上所述,这些数据揭示了USP13的过表达在致癌的Kras激活和Trp53缺失的背景下驱动肺鳞状细胞癌的发展。
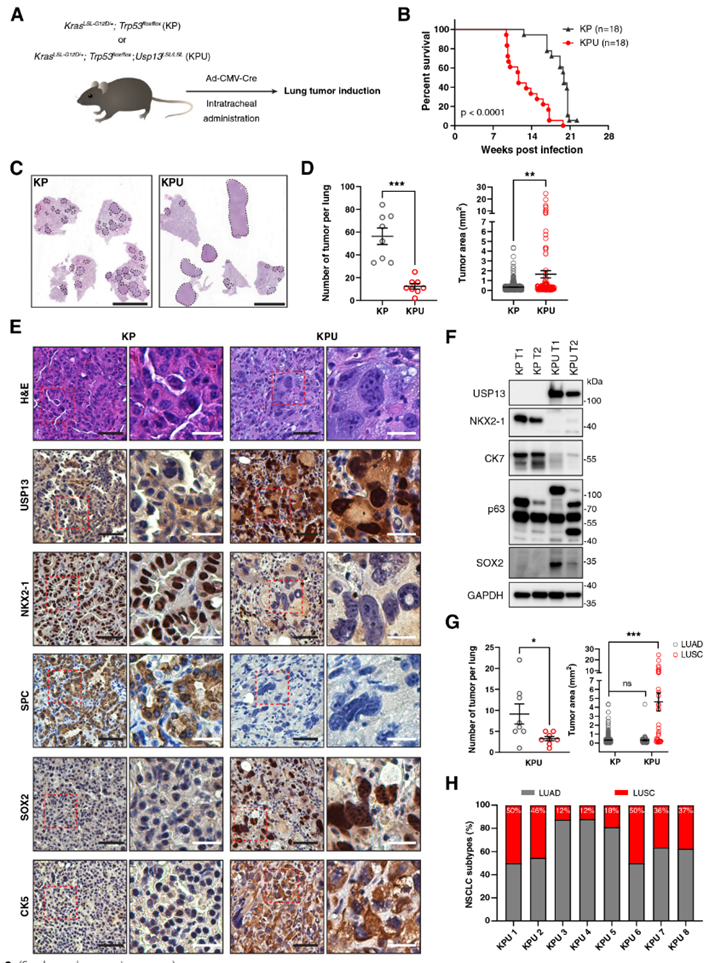
图2 在KP小鼠模型中,USP13过表达驱动肺鳞状细胞癌的发展
3. 谱系重编程通路在KPU肿瘤中富集
为了分析KPU肿瘤的分子特征,进行批量RNA测序。与KP肿瘤相比,KPU肿瘤转录组表现出更大的异质性,并被分为两个不同的簇(KPU 1型和KPU 2型)(图3A)。与组织病理学分析一致,在KPU肿瘤中,肺鳞状细胞癌标记基因的表达水平升高,而肺腺癌标记基因的表达水平下调(图3B)。值得注意的是,KPU 1型肿瘤同时表现出SCLC和LUSC标志基因的表达上调(图3B)。与KP肿瘤相比,KPU肿瘤中有1050个差异表达基因(657个上调,393个下调)。标志性基因集的基因集富集分析显示,KPU肿瘤高度富集于上皮-间充质转化(EMT)通路(图3C)。许多EMT相关基因,如Calu,Fn1,Vim和Snai2,在KPU肿瘤中表达上调(图3D)。
作者进一步发现不同的信号通路在KPU 1型和KPU 2型中富集。Hedgehog和Notch信号通路在KPU 1型中富集,G2/M检查点和PI3K/AKT/mTOR信号在KPU 2型中富集(图3E)。KP和KPU转录组的独创性通路分析(IPA)表明,USP13过表达导致与CREB信号、基底细胞癌信号、干细胞多能性和EMT相关的基因表达改变(图3F)。
在IPA上游调控因子分析中,KPU肿瘤被预测激活由TWIST1,SNAI2,SOX2和MYC转录因子调控的基因网络,这些转录因子驱动EMT,鳞状细胞和谱系重编程(图3G)。另一方面,LUAD谱系特异性转录因子(NKX2-1和FOXA2)预计在KPU肿瘤中是无活性的(图3G)。KPU肿瘤和人肺鳞状细胞癌的比较分析发现,它们共享共同的转录因子,这些转录因子要么是共激活的,要么是失活的(图3G,H)。根据不同的转录组特征,人类肺鳞状细胞可分为4种亚型:原始型、经典型、分泌型和基底型。KPU T1、KPU T3和KPU T4分别表现出与经典、基底和原始人类肺鳞状细胞相似的特征(图3I)。综上所述,KPU肿瘤表现出与人肺鳞状细胞癌相似的分子特征,并显示出强大的肿瘤可塑性和谱系重编程活性,这似乎与鳞状细胞谱系标志物(SOX2)的上调,NKX2-1和FOXA2的下调,以及多能性因子(如MYC,NANOG和KLF4)的激活有关。
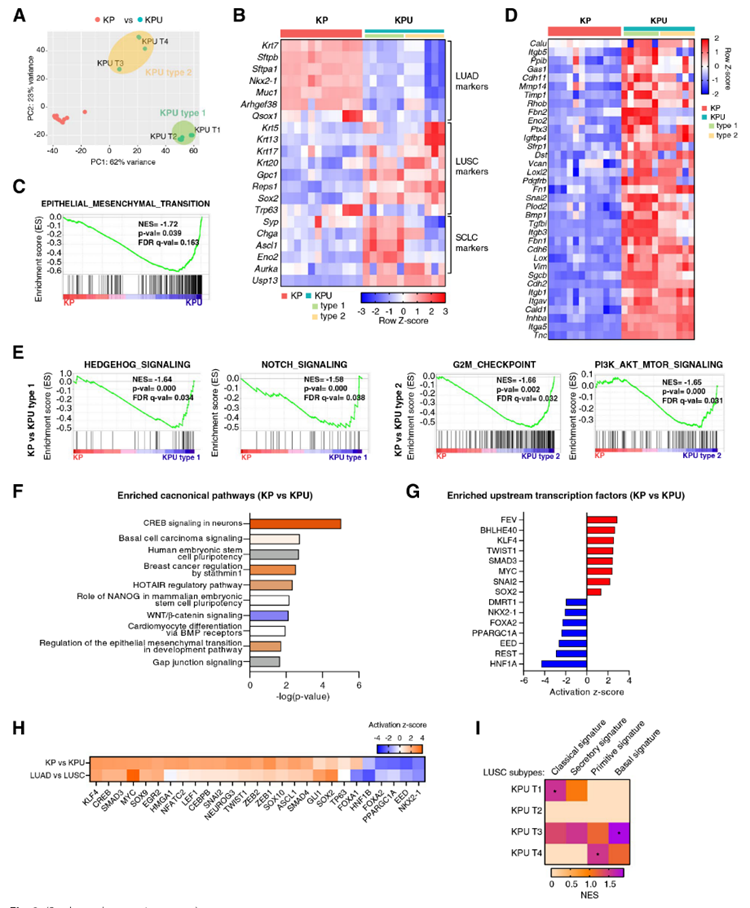
图3 KPU肿瘤的转录组学特征
4. USP13和MYC在人肺鳞状细胞癌和KPU鳞状细胞癌中呈正相关
作者研究了USP13和MYC在人NSCLC中的关系。在人肺鳞状细胞癌临床蛋白质组肿瘤分析联盟(CPTAC)的蛋白质组数据中,USP13、MYC和SOX2在蛋白水平上存在正相关(图4A)。作者进一步使用IHC检测了人NSCLC组织芯片中USP13、MYC和SOX2蛋白的表达。USP13、MYC和SOX2在肺鳞癌组织中的表达显著高于肺腺癌组织(图4B)。MYC和USP13蛋白表达水平在肺鳞状细胞癌和肺腺癌组织中均呈正相关(图4C,D)。
接下来作者研究了MYC在KP和KPU肿瘤中的表达。MYC在KPU肿瘤中强表达,主要在细胞核内(图4E)。特别是,MYC仅在KPU肿瘤的肺鳞状细胞癌成分中升高,而在肺腺癌成分中没有升高(图4F),提示MYC表达的升高可能与KPU肺LUSC的进展有关。建立的原代KPU肺癌细胞MYC表达水平高于KP原代细胞(图4G)。值得注意的是,外源性USP13过表达诱导了多种人NSCLC细胞株中MYC表达水平的上调(图4H)。这些数据揭示了USP13与肺癌中MYC的水平和/或活性相关,并暗示了USP13和MYC在肺鳞癌发展中的潜在作用。
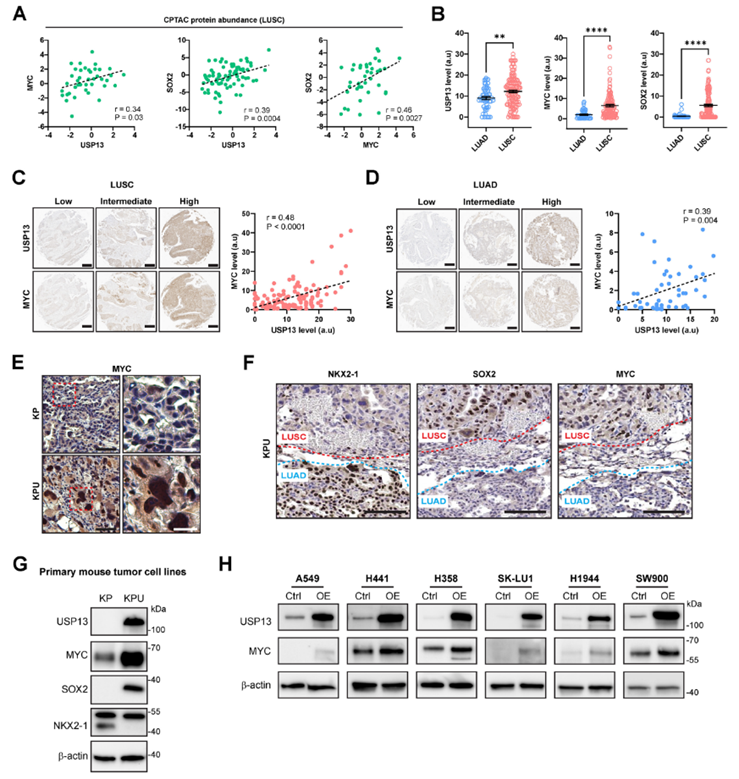
图4 MYC蛋白在肺癌中通过USP13上调
5. USP13通过其去泛素化酶活性稳定MYC蛋白
鉴于USP13在胶质母细胞瘤和肝细胞癌中调节MYC的稳定性,促进自我更新或致瘤潜能,作者假设USP13与肺癌中MYC蛋白丰度直接相关。过表达USP13显著增加MYC在蛋白水平上的表达,但在mRNA水平上没有显著增加,表明USP13对MYC的翻译后调控(图5A)。USP13敲低后,外源性MYC表达水平显著降低(图5B)。免疫共沉淀显示了USP13与MYC的相互作用(图5C)。在放线菌酮追逐实验中,外源性USP13过表达增强了MYC蛋白的稳定性(图5D)。
为了研究USP13的去泛素化酶活性对稳定MYC的影响,作者首先检测了Spautin-1(USP10/USP13去泛素化酶活性的小分子抑制剂)处理的HEK293T细胞中FLAG-MYC的水平。抑制USP13导致FLAG-MYC蛋白丰度降低(图5E)。有趣的是,Spautin-1治疗也导致USP13水平降低。USP13已经描绘了功能域,即一个锌指结构域(ZnF)和两个近端泛素结合结构域(UBA1/2)(图5F)。作者试图确定这些结构域中哪些对MYC的稳定至关重要。USP13的ZnF或UBA1/2结构域的缺失导致其增加MYC蛋白水平的能力下降,而两个结构域的缺失则完全消除了其上调MYC蛋白水平的能力(图5G)。
过表达野生型USP13降低了泛素结合的MYC水平(图5H)。然而,突变体USP13∆Znf∆UBA没有同样的作用(图I)。相反,USP13的敲低增加了MYC的泛素水平(图5J)。为了进一步研究USP13对MYC去泛素化的链特异性,作者将USP13与HA标记的泛素共表达,泛素携带一个位于K48,K63或K48R的赖氨酸残基。免疫印迹数据表明,USP13作用于MYC上的K48连接的泛素链,而不是K63(图5K)。这些结果表明,USP13通过切割MYC的K48连接的泛素结合来增加MYC蛋白的稳定性。
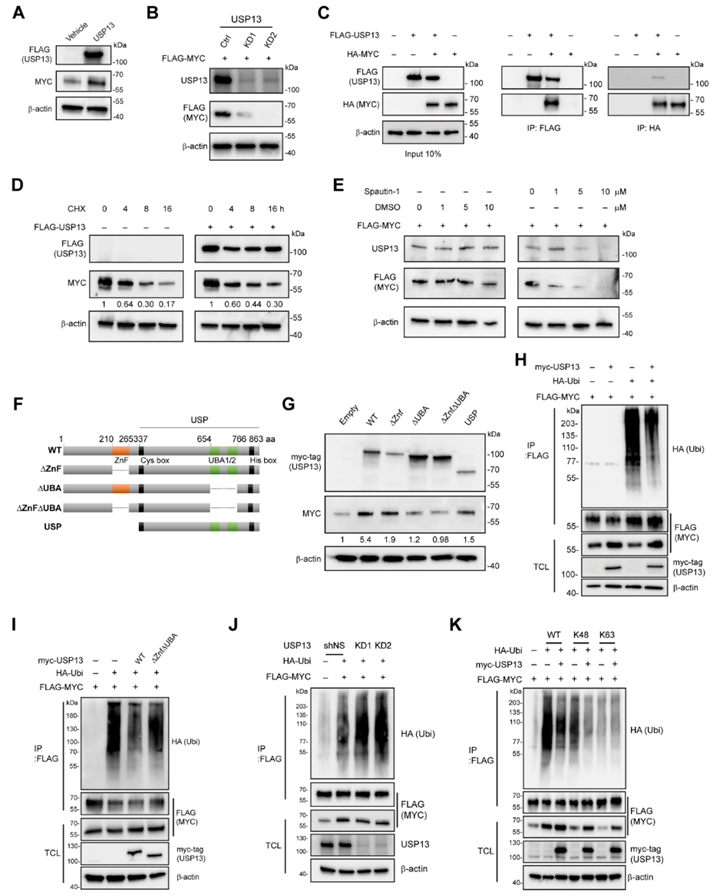
图5 USP13通过其去泛素化酶活性稳定MYC蛋白
6. 在KPU模型中,棒状细胞是肺鳞状细胞癌的起源
为了研究肺祖细胞在KPU驱动的混合LUAD和LUSC肿瘤亚型中的作用,作者通过将Ad5-CC10-Cre或Ad5-SPC-Cre腺病毒分别在KPU小鼠的气管内递送,在棒状细胞和AT2细胞中诱导Cre重组酶的限制性表达(图6A,B)。感染后12~14周,检测两组KPU小鼠的肿瘤负荷和组织病理学。AT2细胞靶向Ad5-SPC-Cre感染的KPU肺显示肺肿瘤结节数量增加,但大小较小,并显示腺癌组织学的存在。当KPU小鼠感染俱乐部细胞中的CC10-Cre病毒时,它们产生了明显较大的具有鳞状组织学特征的肿瘤(图6C)。然后作者分析了肺癌谱系标志物NKX2-1和SOX2的表达。Ad5-SPC-Cre感染的KPU肿瘤大部分NKX2-1阳性,SOX2阴性,只有少数肿瘤NKX2-1阴性(图D,E)。通过Ad5-CC10-Cre检测,KPU肺内大多数肿瘤被鉴定为NKX2-1阴性和SOX2阳性(NKX2-1-/SOX2+),尽管少数肿瘤表现出NKX2-1+/SOX2 -或NKX2-1+/SOX2+的特征(图D,E)。接下来,作者检测了这些因子在Ad5-SPC-Cre或Ad5-CC10-Cre病毒诱导的KPU小鼠肿瘤发展过程中的表达。从早期增生性病变到浸润性癌,AT2细胞来源的肿瘤持续呈现NKX2-1+/SOX2-模式(图6F)。CC10-Cre感染KPU肺后,细支气管增生最初表现为两种因子的阳性表达;然而,NKX2-1的表达随后在原位癌(CIS)阶段下调,在浸润性癌中保持阴性(图6F)。在Ad5-CC10-Cre感染的KPU肺中,NKX2-1水平显著降低,而SOX2水平升高(图6G)。这些数据表明,KPU小鼠的鳞状细胞肿瘤起源于俱乐部细胞,而不是AT2细胞。此外,USP13可能参与了NKX2-1在肺鳞状细胞癌发展早期的下调。
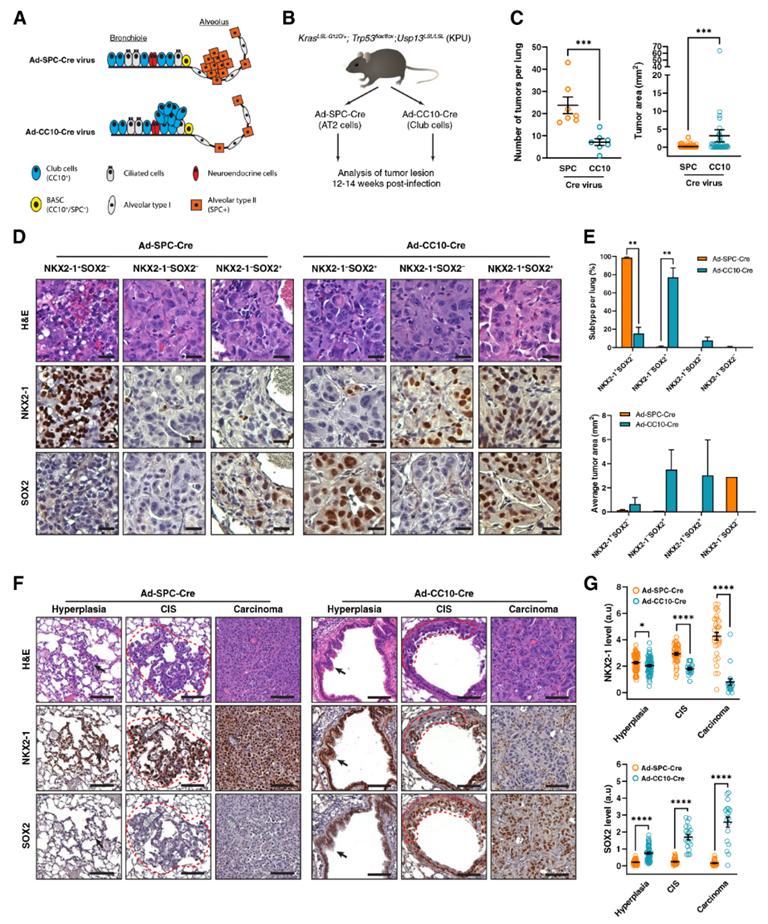
图6 在KPU模型中,LUSC起源于CC10棒状细胞
7. 在棒状细胞来源的肺鳞状细胞癌中,USP13抑制NKX2-1而上调SOX2
AT2细胞和棒状细胞都被认为是KP诱导的LUAD发育的起源细胞。在之前研究中发现,CC10+棒状细胞来源的细支气管增生和腺瘤中SOX2呈阳性表达,但在腺癌中SOX2表达下调,导致LUAD谱系同质。在KPU模型中,在KP遗传改变背景下,棒状细胞中USP13的过表达导致鳞状细胞癌的发展。因此,作者进一步研究了分别递送Ad5-CC10-Cre或Ad5-SPC-Cre病毒后KP和KPU模型的肿瘤进展情况(图7A)。Ad5-CC10-Cre诱导的KPU肺发生了少量的肿瘤病变,但与KP肺相比,肿瘤较大(图7B,C)。正如预期的那样,棒状细胞来源的KP肿瘤呈腺癌组织学,LUAD标志物(NKX2-1和SPC)表达阳性(图7D)。
KPU肿瘤中SOX2的表达水平是KP肿瘤的5.2倍(图7E)。有趣的是,CC10-Cre诱导的KP肿瘤也显示SOX2阳性表达,并且其水平是异质性的(图7E)。在Ad5-CC10-Cre感染的KP和KPU肺中,KP和KPU细支气管中的增生细胞染色显示NKX2-1和SOX2均呈阳性(图7F)。作者的数据表明,KP驱动的增生性病变和由棒状细胞转化的腺瘤均表达SOX2,但在更晚期的病变中,SOX2表达显著降低和部分丢失(图7F,G)。在CIS阶段,KP和KPU小鼠肺组织中NKX2-1和SOX2的表达模式存在明显差异。与KP模型相比,在KPU模型中,NKX2-1的表达在CIS阶段显著降低,而SOX2的表达在整个肺鳞状细胞癌发展过程中显著升高(图7F,G)。
接下来,作者检测了Ad5-CC10-Cre诱导后KP和KPU小鼠肿瘤发展过程中细胞身份标志物CC10和SPC的表达(图7H)。KP和KPU的细支气管增生性病变均表现为CC10+/SPC-。然后,从增生性病变到腺瘤和浸润性腺癌,KP转化的棒状细胞从CC10+/SPC-逐步转变为CC10-/SPC+(图7H)。相反,KPU癌没有显示SPC表达升高,显示了CC10+/SPC-到CC10-/SPC-的不同谱系标志物转换(图7H)。这些数据提示USP13参与细胞谱系重编程,并将致癌性Kras介导的转化的CC10+/SPC-棒状细胞的命运转变为CC10-/SPC-,从而导致鳞状细胞分化而不是腺癌进展(图7I)。
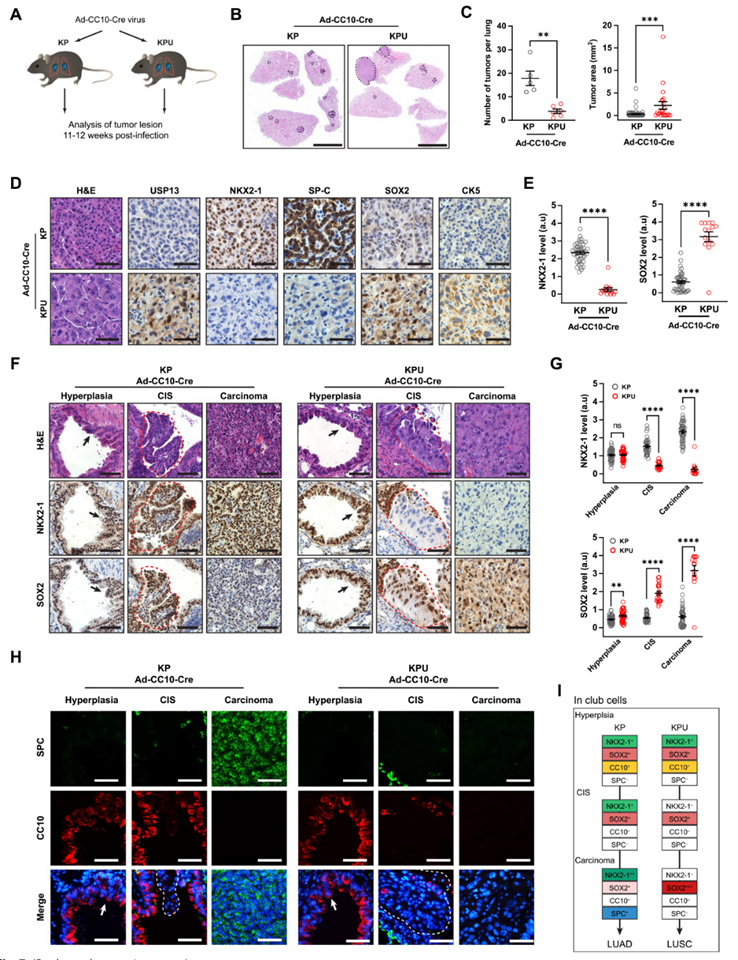
图7 在kp转化的俱乐部细胞中,USP13表达增强可改变谱系因子的表达
8. USP13引起的MYC蛋白水平升高有助于促进鳞状细胞癌的特征
鉴于人肺鳞状细胞癌和KPU肺鳞状细胞癌中USP13和MYC之间的强关联,以及通过USP13的去泛素化活性实现的MYC蛋白的稳定,作者检测了Ad5-SPC-Cre或Ad5-CC10-Cre感染后KP和KPU肿瘤发生过程中MYC的表达模式。无论USP13是否过表达,Ad5KPU肺浸润性鳞状细胞癌的细胞质和细胞核均检测到MYC强表达(图8D)。在Ad5-CC10-Cre感染的KP肺的CIS阶段,MYC+细胞的比例急剧减少,并在腺癌发生过程中继续减少(图8A)。在人类,MYC已被报道主要在肺癌患者气道的正常基底细胞的细胞质中,但细胞质中的MYC转移到癌前病变和鳞状细胞的细胞核中。同样,在Ad5-CC10-Cre感染的KPU小鼠中,MYC在鳞状上皮CIS病变处移位入核,MYC的核强表达在鳞状上皮癌发展过程中持续存在(图8A)。这些发现提示USP13的去泛素化活性可能在肺鳞状细胞肿瘤发生的早期阶段稳定MYC蛋白,并且这种作用仅限于起源于棒状细胞的鳞状细胞肿瘤。
最近的一项研究提出MYC可能是与肺腺癌和肺鳞状细胞癌组织学转化相关的分子驱动因子。为了确定USP13是否可以通过MYC改变晚期肺肿瘤的谱系特征,作者在小鼠腺癌KP原代细胞系和人肺腺癌细胞系中过表达USP13,然后通过免疫印迹法检测鳞状细胞标志物,如SOX2,p40或CK5。单独过表达UPS13可诱导鳞状细胞标志物的表达(图8C,D)。此外,MYC过表达也增加了鳞状细胞标志物的水平(图8C,D),这意味着USP13可以诱导小鼠和人肺腺癌细胞的MYC和鳞状细胞谱系程序,并且MYC足以增强肺腺癌的特征。为了确定MYC是否需要USP13来上调SOX2的表达,作者敲低或抑制MYC,然后在H441细胞中过表达USP13。尽管MYC敲低,过表达USP13仍然导致H441细胞中SOX2水平升高(图8E)。此外,在使用小分子c-MYC抑制剂10058-F4处理的情况下,USP13仍然上调了MYC抑制下SOX2的表达(图8F),表明USP13可能以MYC依赖和MYC非依赖的方式与鳞状细胞标志物相关。根据这些发现,USP13的过表达可能导致起源于棒状细胞的肿瘤发展的初始阶段MYC蛋白的增加,这可能有助于肺鳞状细胞癌的发展。此外,作者发现USP13和/或MYC的过度表达在人类和小鼠肺腺癌中诱导鳞状细胞谱系标记。
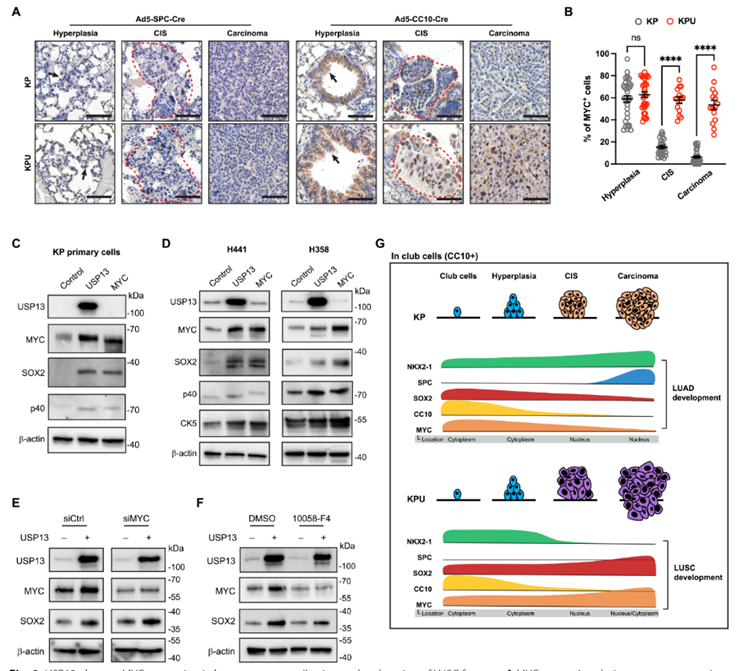
图8 USP13可上调肺癌MYC的表达,促进肺鳞状细胞癌特征的增强
结论:
该研究表明USP13在棒状细胞的鳞状细胞癌变过程中发挥开关作用,并且USP13介导的谱系重编程在细胞起源和遗传背景中得到了至关重要的定义。USP13介导的去泛素化可导致MYC表达上调,这可能为肺鳞状细胞癌的发展和转分化提供了新的分子机制。未来的研究需要确定USP13如何在USP13扩增的肺癌中重编程谱系可塑性,以及靶向USP13是否可以克服治疗耐药性的肺鳞状细胞癌转化的肺肿瘤。
实验方法:
小鼠实验,细胞培养,免疫组化,免疫荧光,免疫印迹,实时荧光定量PCR,RNA-seq,通路富集,免疫沉淀,泛素检测
参考文献:
Kwon J, Zhang J, Mok B, Allsup S, Kim C, Toretsky J, Han C. USP13 drives lung squamous cell carcinoma by switching lung club cell lineage plasticity. Mol Cancer. 2023 Dec 13;22(1):204. doi: 10.1186/s12943-023-01892-x.