






缺乏COL6/VI胶原通过损害自噬和诱导细胞凋亡导致巨核细胞功能障碍
内质网应激在结缔组织疾病的分子病理中起着重要的作用。为了应对内质网应激,细胞可以上调巨噬/自噬,这是细胞降解和回收蛋白质或去除受损细胞器的基本细胞稳态过程。在这些情况下,自噬激活可以支持细胞存活。在这里,我们通过体外和体内方法证明,来自Col6a1-⁄-(胶原,VI型,α1)缺失小鼠的巨核细胞显示COL6多肽的细胞内保留增加,内质网应激和凋亡。未折叠蛋白反应在col6a1-/-巨核细胞中被激活,这可以通过分子伴侣的上调、Xbp1 mRNA剪接的增加和促凋亡调节因子DDIT3/CHOP的高水平来证明。尽管内质网应激,基底自噬在col6a1-/-巨核细胞中受损,表现为BECN1水平降低和自噬体成熟度降低。饥饿和雷帕霉素治疗可挽救巨核细胞的自噬通量,导致细胞内COL6多肽潴留、内质网应激和细胞凋亡减少。此外,Bethlem肌病和Ullrich先天性肌营养不良(两种col6相关疾病)患者外周血造血祖细胞培养的巨核细胞表现出凋亡增加、内质网应激和自噬受损。这些数据表明,巨核细胞中胶原遗传障碍、内质网应激和自噬调节可能是相互关联的。该研究于2023年3月发表于《Autophagy》上,IF=13.3,题为“Lack of COL6/collagen VI causes megakaryocyte dysfunction by impairing autophagy and inducing apoptosis”。
技术路线
研究思路
1. Col6a1-/-Mks中的内质网应激
作者研究了COL6合成的改变是否会影响内质网蛋白合成的稳态。RT-qPCR分析体外培养或直接从BM样品中分选的Mks的UPR通路发现,Col6a1-/-细胞显示编码ATF4(激活转录因子4)和ATF6、PPP1R15A/GADD34(蛋白磷酸酶1,调节亚基15A)、HSPA5/Bip(热休克蛋白5)、HSP90B1/GRP94(热休克蛋白90,β (GRP94),成员1)和胶原特异性伴侣SERPINH1/HSP47(丝氨酸(或半胱氨酸)肽酶抑制剂,分支H、成员1),与野生型(WT)细胞相比,编码XBP1 (X-box结合蛋白1)的转录本剪接增加(图1A-C)。这些数据通过UPR关键调控因子的western blot分析得到证实。此外,Col6a1-/-Mks的特征是DDIT3/CHOP蛋白水平显著升高(图1d和E),这是一种与PPP1R15A激活直接相关的转录因子,它有助于在未解决的慢性内质网应激中激活细胞死亡信号。UPR诱导导致内质网结构改变,从而增加蛋白质折叠能力。共聚焦和透射电镜显示,培养的Col6a1-/-Mks显示内质网扩张,这是内质网应激的典型标志(图1F-H)。通过直接染色Col6a1-/-BM活检获得的Mks证实了相同的形态畸。
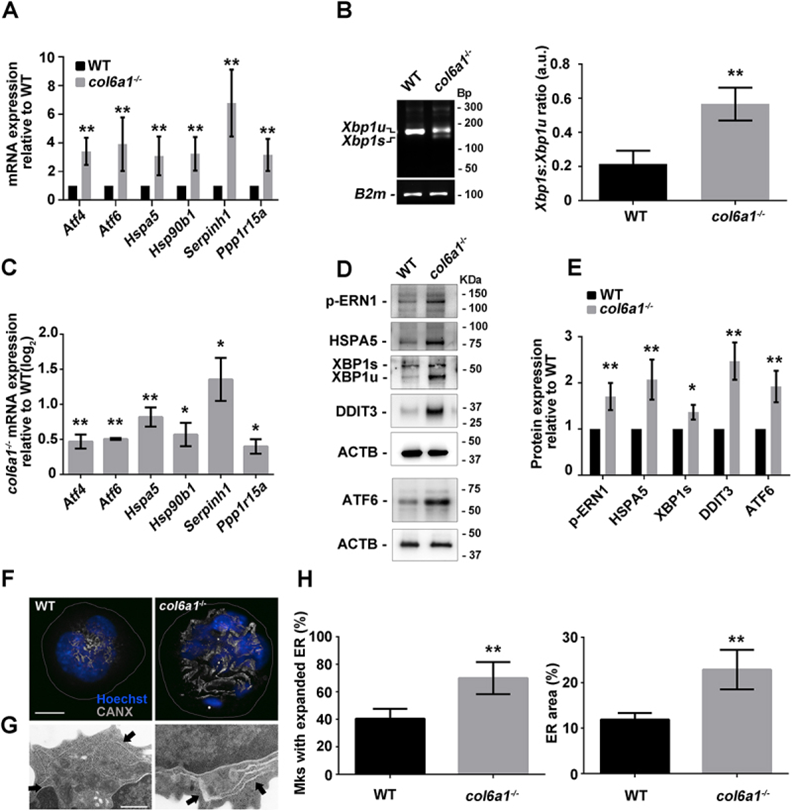
图1 Col6a1-/-Mks内质网应激
2. Col6a1-/-Mks细胞内COL6的积累
与WT相比,在抗坏血酸存在下培养的Col6a1-/-Mks不能分泌COL6,这在培养基中或靠近细胞膜的地方是检测不到的。此外,作者用COL6多克隆抗体共聚焦显微镜和COL6 α2链单克隆抗体western blot分析了COL6在Mks中的存在。这些实验表明,与WT mk相比,Col6a1−/−中COL6链的细胞内保留率显著增加(图2a和B)。
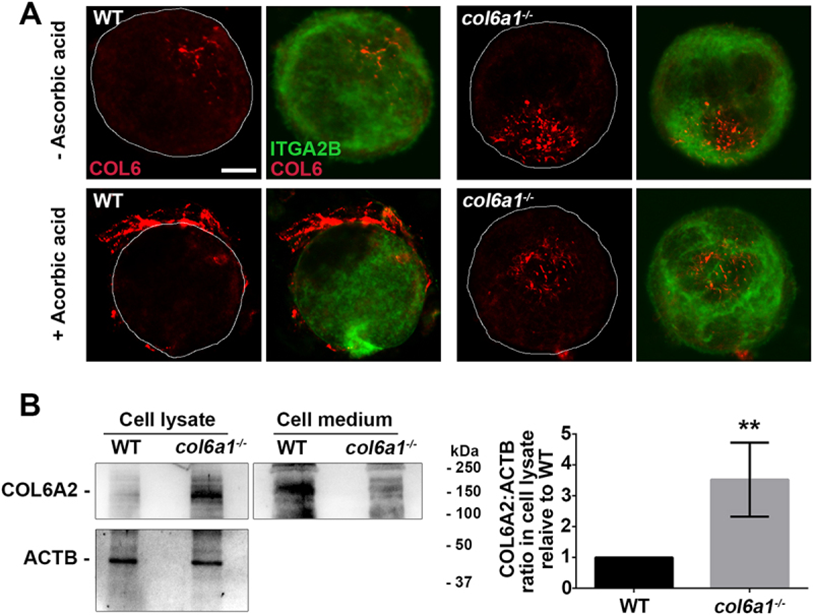
图2 Col6a1-/-Mks细胞内COL6的积累
3. Col6a1-/-Mks的基础自噬激活缺陷
为了深入了解自噬激活状态,Mks被mCherry-EGFP-LC3B报告质粒转染。这种结构可以区分自噬体(mCherry EGFP)和自噬体与溶酶体(即自噬体)融合产生的晚期自噬结构,在自噬体中,由于溶酶体依赖囊泡腔的酸化而发生EGFP破坏(mCherry EGFP+++ -)。Col6a1-/-Mks显示自噬起始减少,LC3B点总数,WIPI2点数量和BECN1 (beclin 1,自噬相关)蛋白水平显示(图3A和B)。此外,Col6a1-/-Mks显示自噬体成熟减少,mCherry EGFP + - LC3B点的数量较低,仅mCherry点的百分比较低,表明基础自噬通量减弱(图3A-C)。这些数据通过使用溶酶体抑制剂氯喹处理Mks得到证实,随后进行LC3B和SQSTM1/p62周转率的western blot分析(图3D和E)。
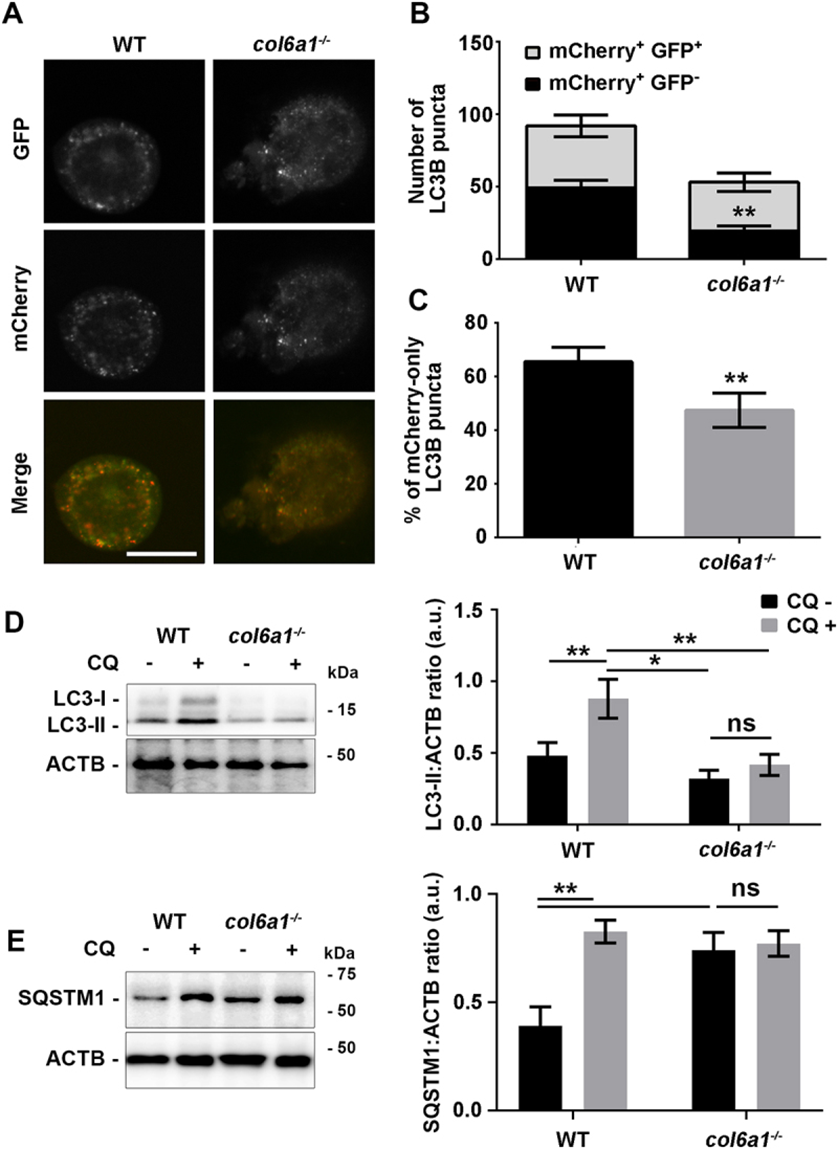 图3 Col6a1-/-Mks的基础自噬激活不足
图3 Col6a1-/-Mks的基础自噬激活不足
4. Col6a1-/-小鼠Mks和血小板凋亡增加
为了研究UPR的诱导和基础自噬通量的改变是否影响Mk凋亡,作者用ANXA5/annexin V和7-氨基放线菌素D (7-AAD)染色WT和Col6a1-/-Mks,测定细胞凋亡/坏死率。在体外和体内,更高比例的Col6a1-/-Mks发生凋亡(ANXA5/annexin V 7-AAD+ -),最终导致更高比例的坏死Mks (ANXA5/annexin V 7-AAD++)(图4A和B)。与WT相比,Col6a1-/-Mks中促凋亡蛋白BAX与抗凋亡蛋白BCL2和BCL2L1/Bcl-xL的比例增加[21]证实了这些数据(图4C和D)。与WT相比,Col6a1-/-小鼠的外周血血小板中ANXA5/annexin V+的比例更高(图4E和F), BAX与BCL2和BCL2L1/Bcl-xL的比例增加(图4)。因此,Col6a1-/-小鼠的体内血小板在外周血中被过早清除,t1/2减少了约30%(从大约1/2)。60 h至40 h),通过跟踪生物素标记的血小板的体内存活情揭示了这一点。为了证明Col6a1-/-小鼠中观察到的血小板缺陷是否是血小板固有的,作者通过在Col6a1-/-小鼠中注入WT血小板和在WT小鼠中注入Col6a1-/-血小板进行了互惠过继转移。通过这种方法,作者发现WT和Col6a1-/-外周血血小板都保持了原来的半衰期,从而表明半衰期的减少是血小板固有的,而不依赖于环境因素。
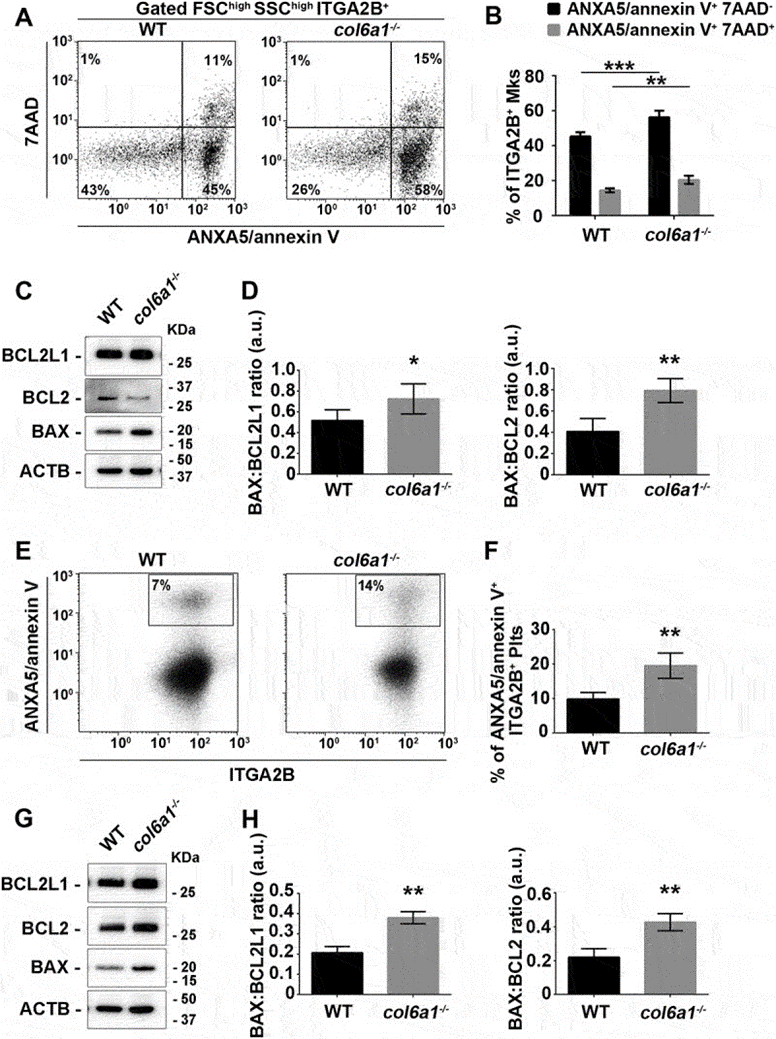
图4 Col6a1-/-Mks和血小板在体外和体内显示出增加的凋亡
5. 饥饿和雷帕霉素处理可挽救Col6a1-/-Mks的自噬激活
自噬受PI3K-AKT-MTOR通路调控,作者最近证实该通路在Col6缺乏的Mks中被过度激活。为了避免Col6a1-/-Mks显示的基础自噬诱导和自噬通量的减少,作者进行了血清饥饿和雷帕霉素治疗,这是两种已知的自噬刺激[23-25]。两种处理都恢复了MTOR通路的激活,并同时诱导了Col6a1-/-Mks中的自噬和自噬通量,这可以通过LC3B点的总数、mCherry EGFP+ -点的数量和仅mCherry点的百分比来证明(+图5 A-C)。在血清饥饿和雷帕霉素治疗下,用氯喹阻断溶酶体功能导致Col6a1-/-Mks中LC3B脂化相对WT进一步增加(图5D)。这些处理也挽救了自噬底物SQSTM1/p62的积累(图5E),并诱导了Col6a1-/-Mks中WIPI2点的形成。
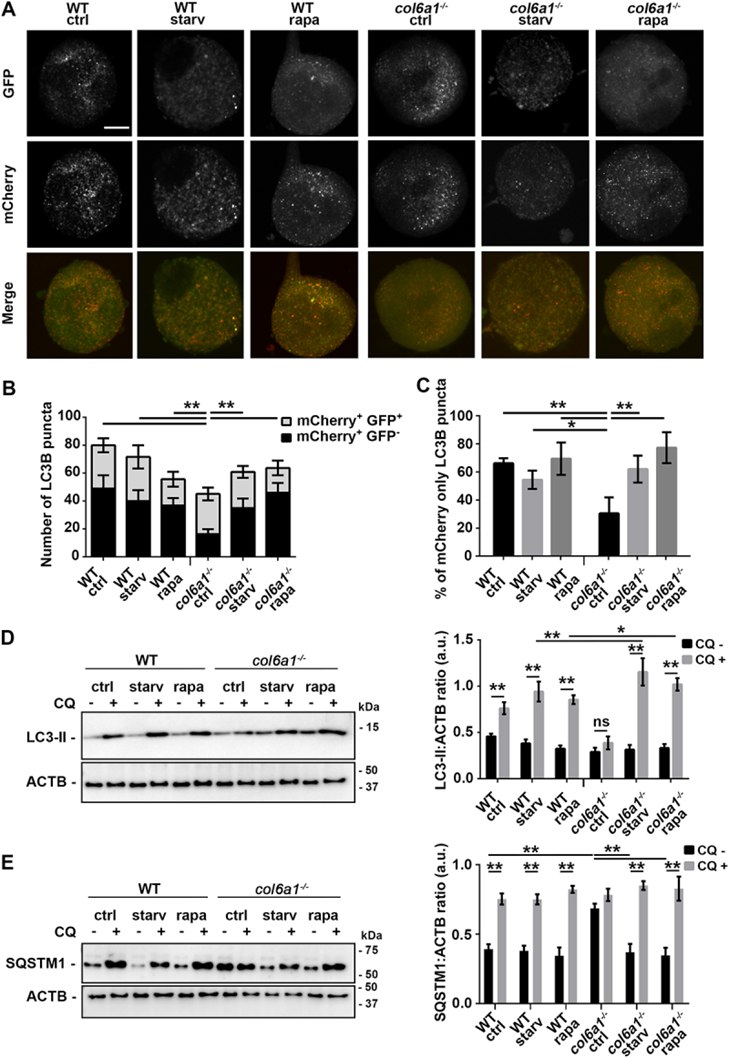
图5 饥饿和雷帕霉素治疗可挽救Col6a1-/-Mks的自噬激活
6. 饥饿和雷帕霉素处理可挽救Col6a1-/-Mks细胞内COL6滞留、内质网应激和细胞凋亡
饥饿和雷帕霉素处理增加了WT和Col6a1-/-Mks中lc3b阳性自噬体和lamp2阳性溶酶体与内质网的共定位(图6A-F),随后Col6a1-/-Mks中细胞内COL6潴留量恢复到WT水平,共聚焦显微镜显示(图7A和B))。western blot分析培养基中分泌的COL6表明,饥饿和雷帕霉素处理后细胞内COL6潴留的减少不是由于COL6的分泌。在bm衍生的Col6a1-/-Mks中,UPR通路在mRNA和蛋白质水平上的显著减少与这种效应相平行(图7C-E)。透射电镜显示,血清饥饿和雷帕霉素处理后,Col6a1-/-Mks未观察到内质网形态学改变(图7F)。为了证明饥饿和雷帕霉素治疗后UPR激活的恢复是由于自噬激活,作者用自噬抑制剂氯喹治疗Mks。在氯喹存在的情况下,两种处理均未能使UPR标记物的表达正常化。血清饥饿和雷帕霉素处理对自噬诱导和内质网应激的影响也对Col6a1-/-Mks的存活有显著影响。事实上,7-aad染色的流式细胞术分析和BAX与BCL2和BCL2L1/Bcl-xL比例的western blot分析显示,血清饥饿或雷帕霉素处理的Col6a1-/-Mks细胞凋亡和细胞死亡显著减少(图7 G-J)。与未处理的对照组相比,体内雷帕霉素处理显著降低了Col6a1-/-bm分类的Mks的内质网应激和自噬分子特征的诱导(图7K和L)。正如作者最近发表的,体内给药雷帕霉素也仅在Col6a1-/-小鼠中诱导循环血小板数量显著增加,达到WT水。
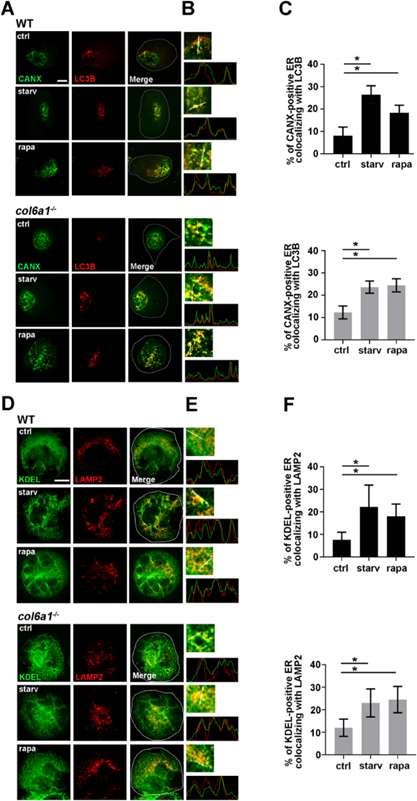
图6 在WT和Col6a1-/-Mks中,内质网与lc3b阳性自噬体和lamp2阳性溶酶体共定位
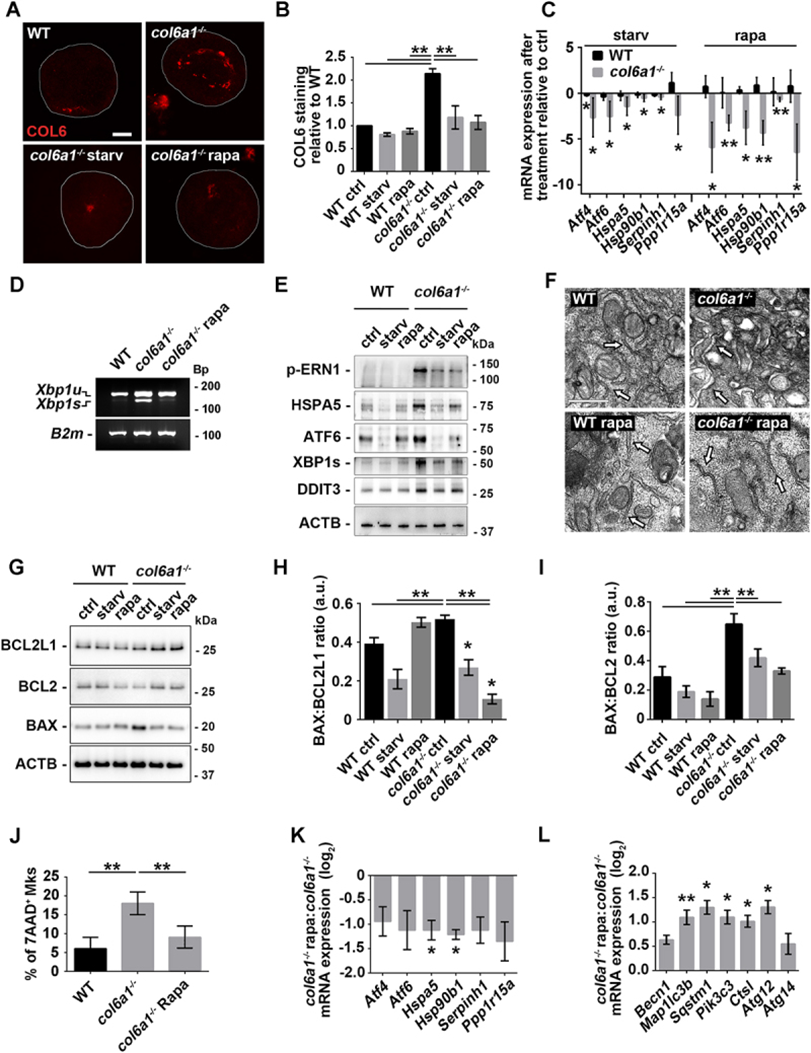
图7 饥饿和雷帕霉素处理可挽救Col6a1-/-Mks细胞内COL6滞留、内质网应激和凋亡
7. Col6相关疾病患者的Mks和血小板中细胞凋亡、内质网应激和自噬缺陷增加
作者从伯利恒肌病和UCMD患者的外周血以及健康受试者的外周血中分化出Mks。来自患者的Mks在分子和蛋白质水平上均显示内质网应激标志物的表达增加(图8A-C))。此外,共聚焦显微镜显示患者Mks的ER明显扩张(图8D)。与来自健康受试者的Mks相比,来自患者的Mks在基础条件下和使用氯喹阻断溶酶体功能后显示LC3B点的数量减少(图8E)。
最后,与健康对照者分化的Mks相比,患者分化的Mks表现出更多的凋亡和坏死(图8F)。与健康受试者相比,患者外周血血小板的细胞凋亡率也较高(图8G)。
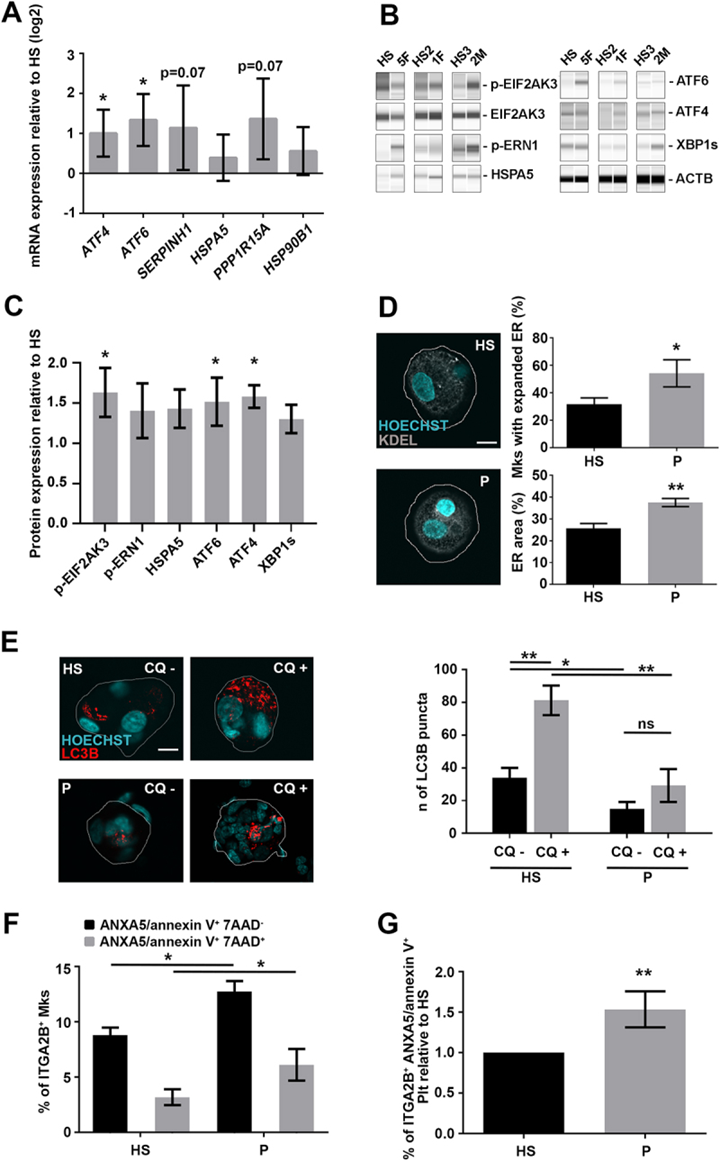
图8 来自Col6相关疾病患者的Mks表现出细胞凋亡增加、内质网应激和自噬缺陷
实验方法
细胞培养;动物建模;RT-qPCR;Western blotting;细胞转染;免疫荧光;小动物成像;流式细胞术;
参考文献:
Abbonante V, Malara A, Chrisam M, Metti S, Soprano P, Semplicini C, Bello L, Bozzi V, Battiston M, Pecci A, Pegoraro E, De Marco L, Braghetta P, Bonaldo P, Balduini A. Lack of COL6/collagen VI causes megakaryocyte dysfunction by impairing autophagy and inducing apoptosis. Autophagy. 2023 Mar;19(3):984-999. doi: 10.1080/15548627.2022.2100105. Epub 2022 Jul 20. PMID: 35857791; PMCID: PMC9980446.