






口腔癌患者的放化疗影响唾液微生物群
口腔鳞状细胞癌(SCC)与口腔微生物生态失调有关。在这项独特的研究中,作者通过16S rRNA基因测序比较了SCC患者治疗前后的唾液微生物组,并检查了微生物组学变化与抗微生物蛋白表达的相关性。SCC的治疗与总体细菌丰富度和多样性降低有关。微生物群落结构发生了显著变化,包括卟啉单胞菌科和普雷沃菌科的丰度下降,乳酸杆菌科的丰度增加。在放化疗治疗前后,微生物群落结构也发生显著变化,而仅手术治疗没有发生变化。在单独接受放化疗的患者中,在治疗前后,有反应者和无反应者之间有几种细菌种群的丰度存在差异。微生物组学的变化与DMBT1(一种人类唾液中的抗微生物蛋白)的表达变化有关。此外,作者发现,治疗后增加的唾液DMBT1可以作为治疗后唾液生物标志物,这与微生物变化有关。具体而言,治疗后人类唾液DMBT1的增加与孪生球菌属spp.、巴斯德氏菌科spp.、乳酸菌spp.和 奥利杆菌属spp.丰度增加相关。这是首次研究SCC患者口腔微生物组学治疗相关变化(放化疗和手术)以及唾液中抗微生物蛋白表达变化的纵向研究。口腔微生物群的组成可以预测治疗反应;唾液DMBT1可能在调节SCC患者的口腔微生物组学中发挥作用。本研究于2023年11月发表在《Microbiome》,IF 15.5。
技术路线:
主要研究结果:
1 唾液微生物组学的纵向分析
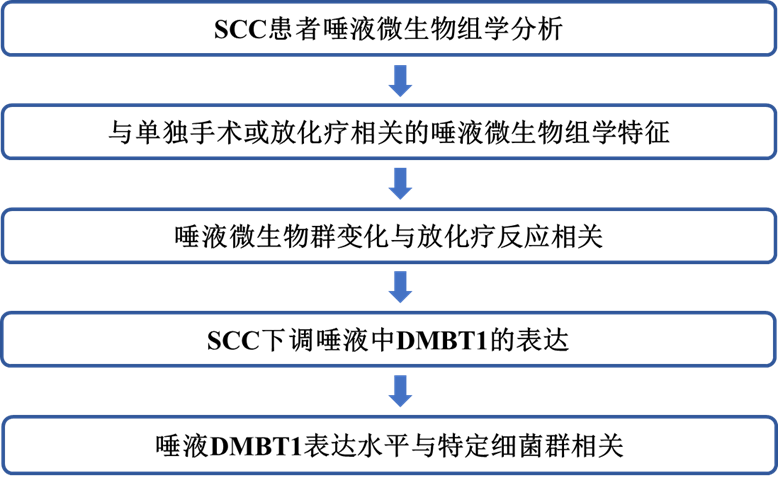
图1A是整个研究工作流程的示意图,包括样本收集、分析和主要比较。Z作者研究了从治疗前到治疗后口腔微生物组学的变化,这些唾液是从SCC患者诊断时和6个月后(0和6个月中)收集的,以确定治疗完成后不久发生的变化。初始治疗包括放化疗、化疗、放疗和手术。NMDS分析表明,在0个月和6个月之间,微生物群落结构存在显著差异(图1B)。此外,治疗处理后样品中的微生物丰富度和多样性均降低(图1C、D)。治疗后厚壁菌门显著增加,拟杆菌、梭杆菌和变形菌门减少(图1E)。在相对丰度至少为0.1%的细菌家族中,拟杆菌科、伯克霍尔德菌科、丹毒菌科、黄杆菌科、毛螺菌科、奈瑟菌科、乳杆菌科、钩端菌科、普雷沃菌科、巴氏杆菌科、消化链球菌科和卟啉单胞菌科显著减少,双歧杆菌科、乳细菌科和假单胞菌科明显增加(图1F)。有几种OTU在治疗后显著降低,包括未分类的巴氏杆菌科(OTU0006)、卟啉单胞菌属(OTU0028,Lachnoanaerobaculum(OTU0045)、未分类的黄杆菌科(OTU0064)、未归类的乳杆菌属(OTU0013)和普雷沃氏菌(OTU0002)(图1G)。该分析中只有两个OTU在治疗后显著增加,即链球菌(OTU0024)和乳酸杆菌(OTU0025)。
通过LEfSe分析,其中一些菌在0月至6个月之间差异最大,尽管发现其他OTU与基线样品相关,包括放线菌(OTU0007)和普雷沃氏菌(OTU4010),或在治疗后样品中富集,包括乳杆菌(OTU0011)、普雷沃氏菌,乳酸杆菌(OTU0020)和假单胞菌(OTU0066)。总之,这些研究表明,SCC治疗后唾液微生物组学发生了变化。
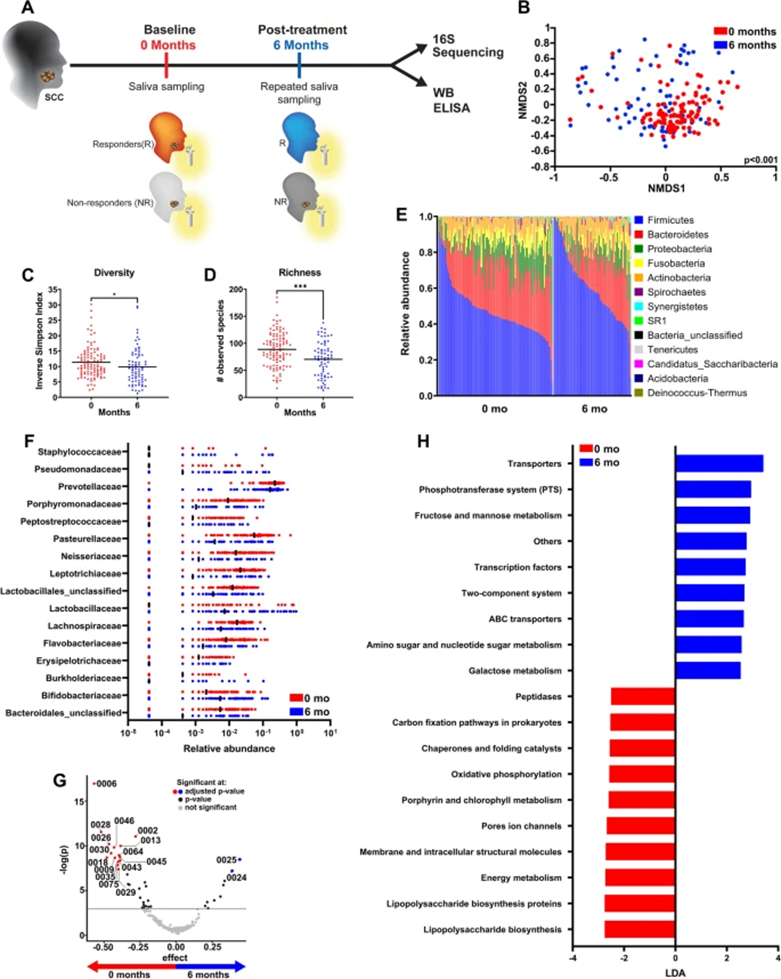
图1:SCC治疗后口腔微生物组学的多样性和丰富度下降
2 与单独手术或放化疗相关的唾液微生物组学特征
放疗、手术、放化疗和化疗是SCC的治疗选择。由于微生物组学可以影响治疗反应,使用从SCC患者第0个月和第6个月采集的匹配样本,作者研究了与放化疗(图2)或手术(图3)相关唾液中的微生物变化。由于样本量小(<12/组),排除了接受手术后辅助放疗、手术后放化疗或仅接受放疗或仅接受化疗的患者。
与开始治疗前的时间0相比,接受0个月和6个月放化疗的匹配样本的患者(n=33)在6个月时表现出微生物群落结构的显著差异(图2A)。多样性没有显著变化(图2B),尽管治疗后丰富度下降(图2C)。在门水平上,厚壁菌门的相对丰度增加,拟杆菌门和变形杆菌门相对丰富度减少(图2D)。此外,放化疗后,在丰度>0.1%的科中,普雷沃菌科、巴氏杆菌科、奈瑟菌科、钩端螺旋菌科和钩端螺旋杆菌科显著减少,乳杆菌科和假单胞菌科增加。ALDEx2进一步鉴定与治疗前和治疗后相关的OTU。具体而言,乳酸杆菌(OTU0025)显著增加,未分类的巴氏杆菌科(Pasteudellaceae spp.)显著减少。通过LEfSe分析,其中一些OTU在放化疗前后也被鉴定为具有差异性丰富。PICRUST分析显示,参与膜和细胞内结构分子、能量代谢、细菌分泌系统和细菌运动蛋白的通路在治疗前具有更高的相关性;参与转运蛋白和苯丙氨酸、酪氨酸和色氨酸生物合成通路的代表性增加与治疗后有关(图2H)。总之,这些研究表明,放化疗治疗SCC后,唾液微生物组学发生了变化。
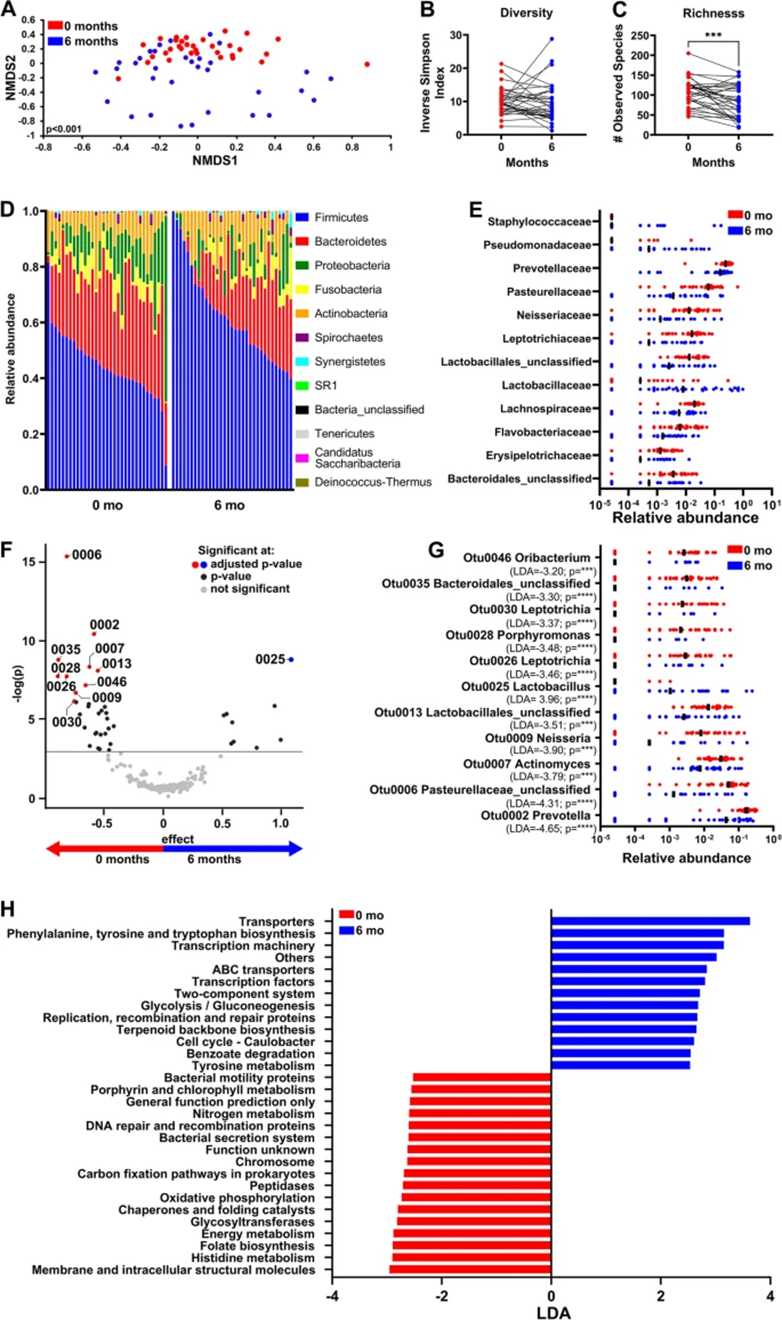
图2:放化疗后微生物组学显著变化
对于单独接受手术治疗的患者(n=15),总体群落结构和多样性没有显著差异(图3A,3B)。有趣的是,丰富度在6个月时显著减少(图3C)。尽管在门(图3D)或家族水平(数据未显示)上的相对丰度在手术前后没有显著差异,但LEfSe分析显示,治疗前未分类的乳杆菌属(OTU0013)、钩端毛滴虫(OTU0106)、未分类的黄杆菌科(OTU0064)以及治疗后的韦氏菌(OTU0185)和假单胞菌(OTU4066)占主导地位(图3E,F)。
基于PICRUSt和LEfSe分析,与治疗相关的KEGG通路显示,与遗传信息处理相关通路,如手术前的翻译(即核糖体)、脂多糖生物合成和DNA复制,以及治疗后涉及氨基酸代谢(D-丙氨酸)通路的代表性增加(图3G)。
图3:单独手术后唾液微生物组学丰富度的显著变化
3 唾液微生物群变化与放化疗反应的关系
确定指示反应的生物标志物具有重要临床意义。收集患者放化疗前(n=50)和放化疗后(n=33)的唾液样本,用于鉴定与放化疗反应相关的潜在微生物生物标志物。如果在随访期间(n=11)出现任何复发(局部或转移),则认为患者无应答,而应答者在最长随访时间之前被认为无疾病(长达5年,n=39)。
在基线(0个月),细菌群落之间的θYC距离显示出总体细菌特征的显著差异(图4A),但在应答者和无应答者之间的多样性(图4B)和丰富度(图4C)没有差异。应答者和无应答者在开始放化疗之前,属于不同门细菌的相对丰度相似(图4D)。此外,无应答者和应答者之间,唾液中丰度大于0.1%的任何观察到的细菌家族的相对丰度均无显著差异(数据未显示),包括先前与口腔癌症相关的家族,如卟啉单胞菌科、普雷沃菌科、链球菌科,或梭杆菌科(图4E)。尽管ALDEx2分析在基线时没有预测到任何与治疗反应显著相关的OTU,但基线唾液样本的LEfSe分析显示,无应答者与普雷沃氏菌(OTU0029)之间存在显著关联(图4F),而未分类的巴氏杆菌科(OTU0006),和劳特罗普氏菌属(OTU0092)在应答者中更为丰富(图4G)。PICRUSt分析未揭示任何与反应或复发相关的预测功能途径(数据未显示)。
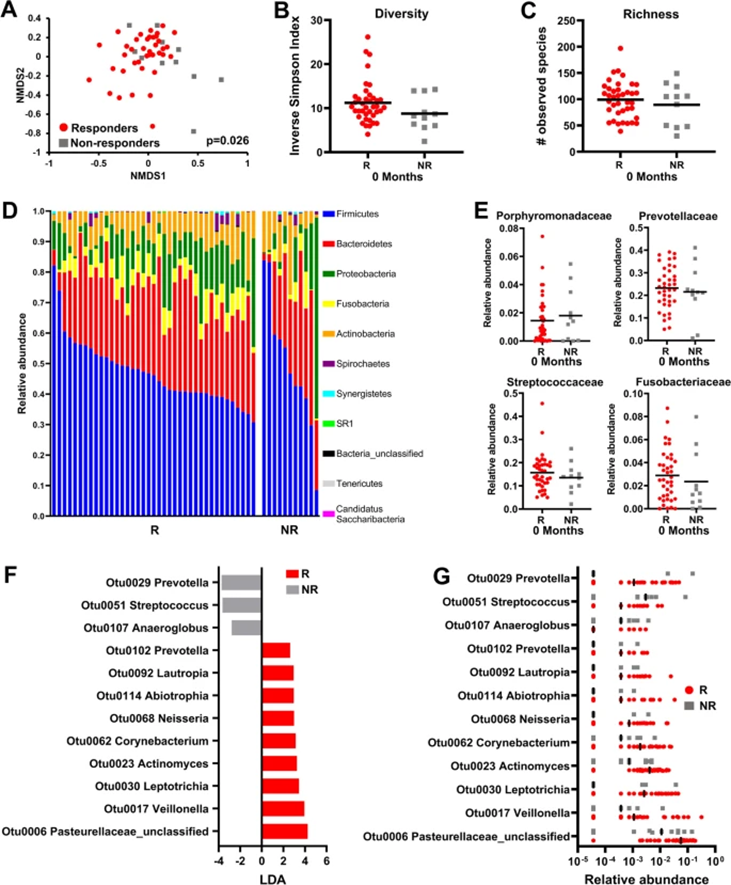
图4:普雷沃氏菌与基线放化疗无反应相关
在6个月完成治疗后,当比较无应答者(n=8)和应答者(n=25)之间的唾液微生物群落时,基于θYC距离的总体群落结构存在更高的显著差异(图5A),但总体多样性(图5B)或丰富度(图5C)没有差异。治疗后,应答者和无应答者在厚壁菌门、拟杆菌门和梭杆菌门中的细菌丰度也相似(图5D),与治疗前应答者和无应答者的唾液微生物群一样,通常与口腔癌菌群失调相关的不同细菌家族的相对丰度没有显着差异,包括卟啉单胞菌科、普雷沃氏菌科或梭杆菌科。然而,观察到链球菌科显着减少(图5E和数据未显示)。治疗后样本的LEfSe分析显示,应答者与韦氏菌(OTU0001)、链球菌(OTU0004)、罗氏菌属(OTU0015和OTU0016)、孪生球菌属(OTU0014)、奇异菌属(OTU0021)和放线酵母(OTU0023)的相对丰度之间存在显著关联(图5F,G)。
由于微生物代谢产物会影响对治疗的反应,因此进行PICRUSt预测从16s rRNA序列推断出的可能与反应相关的功能通路。与治疗前样本不同,治疗前样本在应答者和非应答者之间的预测代谢途径没有差异(数据未显示),治疗后样本显示应答者与氨基酸生物合成和代谢相关途径的代表性增加(图5H)。另一方面,无应答者参与糖代谢、酪氨酸代谢和脂肪酸生物合成的多种途径增加(图5H)。
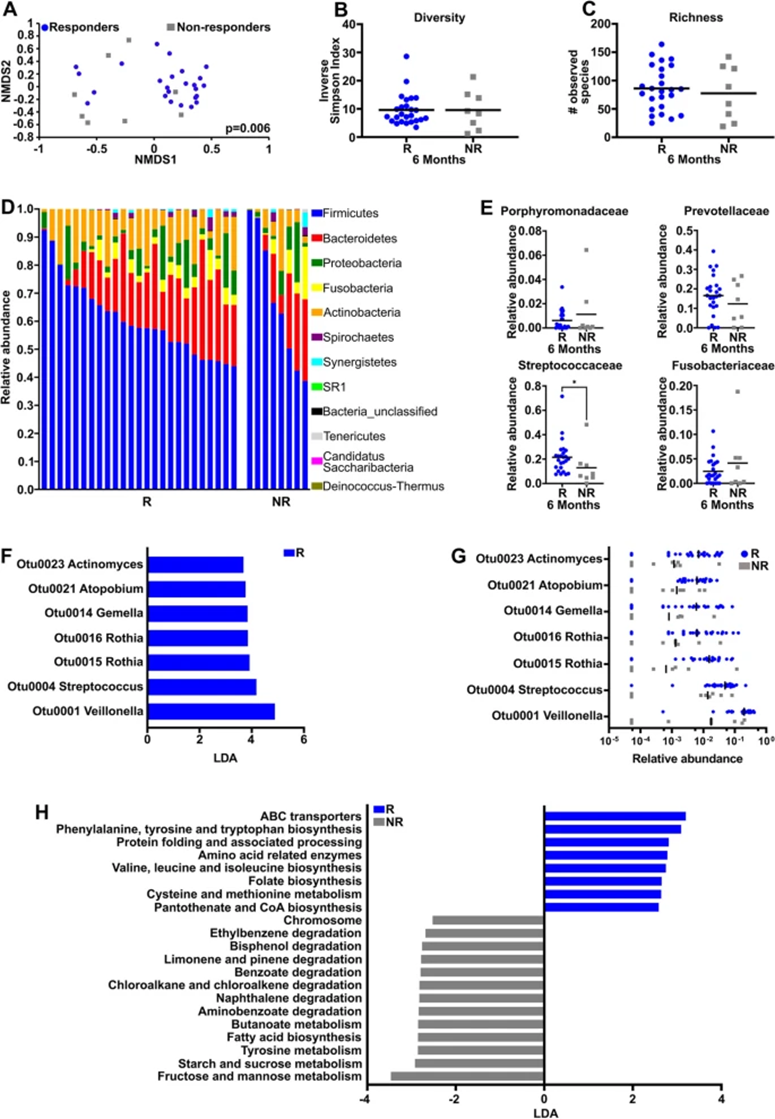
图5:6个月时放化疗应答者和无应答者之间的微生物组学差异
4 SCC下调唾液中的DMBT1
作者之前证明,DMBT1是一种抗微生物蛋白,在SCC中下调,并与侵袭能力增加和预后不良有关。DMBT1在唾液腺中强烈表达,占唾液分泌蛋白的10%。为确定SCC患者唾液中DMBT1的表达是否随治疗而变化,对一组患者进行免疫印迹分析(图6A)。来自48个SCC患者唾液样本的免疫印迹分析显示,与体积标准化样品的基线相比,治疗前DMBT1表达低,治疗后6个月(平均2.1倍)和12个月(1.9倍)水平显著增加(图6B,D)。ELISA (图 6C,E) 对通过免疫印迹分析筛选的 48 名患者中一个子集 (n=28) 的唾液样本观察到类似的发现。无论治疗方案如何,这些数据表明DMBT1表达在SCC治疗后均增加。
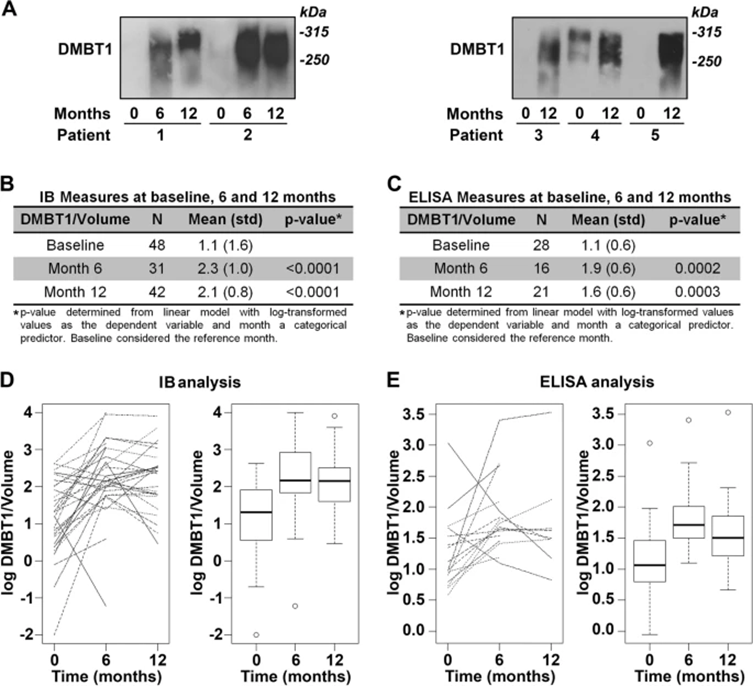
图6:未经治疗的SCC患者唾液中DMBT1分泌受到抑制
为直接研究SCC调节唾液DMBT1分泌的程度,将UM-SCC-1细胞皮下注射到小鼠中,并在两个时间点对成年小鼠唾液中的DMBT1进行定量(图7A)。将DMBT1标准化为唾液体积,以适应小鼠之间分泌的变化。从注射后10天开始,测量肿瘤大小并计算肿瘤体积(图7B)。苏木精-伊红染色和细胞角蛋白免疫组织化学证实了SCC的存在(图7C)。患有肿瘤的成年小鼠的DMBT1分泌显著减少,而对照小鼠的DMBT1分泌在两个时间点之间没有显著差异(图7D,E)。在本实验的线性混合模型中,组x时间的相互作用项趋于显著性。人类和小鼠唾液研究共同表明,SCC抑制唾液中DMBT1的表达。
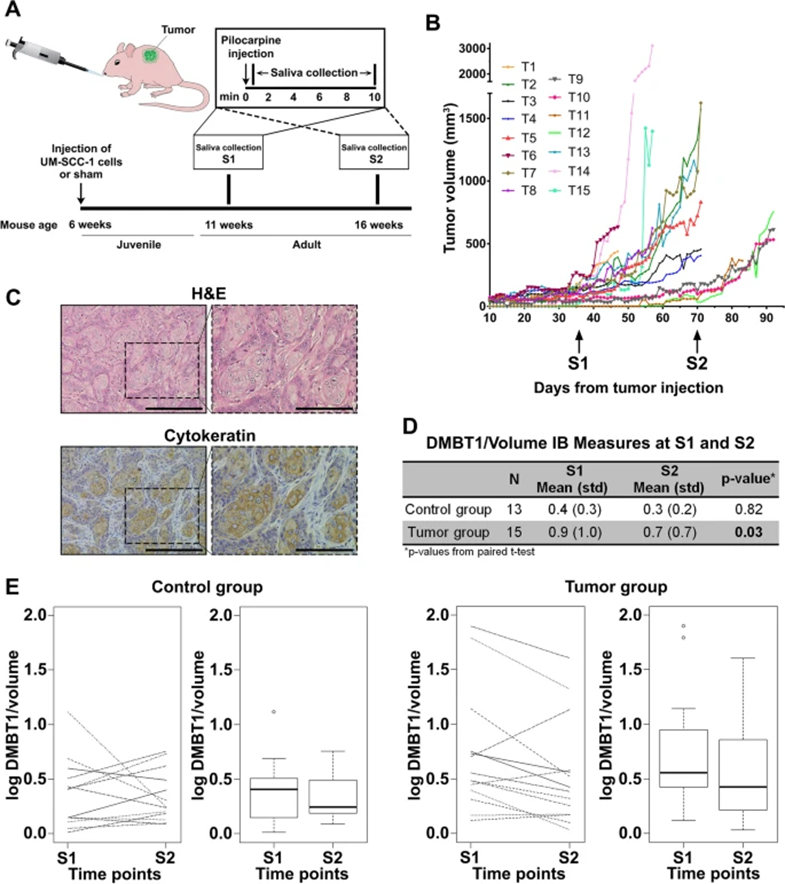
图7:肿瘤发生后小鼠唾液DMBT1减少
5 唾液中DMBT1水平与特定细菌群相关
在人类唾液样本中(图6),作者应用Pearson相关模型研究DMBT1蛋白表达与治疗前后唾液微生物组学组成之间的关系(图8)。治疗前,较低水平的DMBT1与较高的梭杆菌(OTU0065)、未分类的钩体螺科(OTU0072)和未分类的糖念珠菌属(OTU0205)的相对丰度以及较低的密螺旋体(OTU0153和OTU0980)、链球菌(OTU0284)和普雷沃氏菌(OTU496)的相对丰富度相关。治疗后,高DMBT1水平与放线菌(OTU0023和OTU0143)、埃肯菌属(OTU0091)、嗜二氧化碳噬细胞菌(OTU0043和OTU2071)、乳酸杆菌(OTU0131)和链球菌(OTU0024和OTU4624)的丰度呈负相关,而DMBT1表达与未分类厚壁菌门成员(OTU0146),未分类丛毛单胞菌科(OTU0355)、未分类毛螺菌科(OTU0072)、普氏菌属(OTU0087)和Stomatobaculum(OTU0080)的丰度呈正相关(图8)。作者还分析了DMBT1表达随时间变化与OTU丰度变化之间的相关性。有趣的是,治疗后唾液中DMBT1表达的增加与孪生球菌属(OTU0014),其在6个月放化疗的应答者中也富集(图5G),未分类的巴氏杆菌科属(OTU0006),其在0个月时的应答者中富集,乳杆菌(OTU0025),巨球藻(OTU0012)和金杆菌(OTU4046)丰度的增加相关(图8)。
总之,这些发现表明SCC患者口腔微生物组学的变化与唾液抗微生物蛋白DMBT1的表达变化有关。

图8:唾液中DMBT1的下调与微生物组学变化有关
结论:
这是首项纵向研究,旨在调查SCC患者口腔微生物组学的治疗相关差异,并将其与DMBT1表达的变化联系起来。作者的研究结果支持SCC唾液生物标志物和微生物组学生物标志物的发展,以预测对治疗的反应。未来的研究应针对与反应相关的候选微生物物种,如孪生球菌属spp.和纤毛菌属spp.,包括其对SCC治疗反应的表征和潜在机制的鉴定。
实验方法:
DNA提取与扩增,16S rRNA测序,ELISA,WB
参考文献:
Medeiros MC, The S, Bellile E, Russo N, Schmitd L, Danella E, Singh P, Banerjee R, Bassis C, Murphy GR 3rd, Sartor MA, Lombaert I, Schmidt TM, Eisbruch A, Murdoch-Kinch CA, Rozek L, Wolf GT, Li G, Chen GY, D'Silva NJ. Salivary microbiome changes distinguish response to chemoradiotherapy in patients with oral cancer. Microbiome. 2023 Nov 30;11(1):268. doi: 10.1186/s40168-023-01677-w. PMID: 38037123; PMCID: PMC10687843.