






CTNNB1突变型肝细胞癌中靶向MMP9 可恢复CD8+ T细胞介导的抗肿瘤免疫并提高抗PD-1功效
在肝细胞癌(HCC)中,功能增强(GOF)CTNNB1突变(CTNNB1GOF)引起明显的免疫逃逸,并对抗PD-1产生抗药性。在这里,我们的目标是调查CTNNB1GOF HCC介导的免疫逃逸机制,并提出一种新的治疗策略,以增强在HCC中抗PD-1的疗效。进行RNA测序以识别与免疫逃逸相关的CTNNB1GOF的关键下游基因。利用体外共培养系统、小鼠皮下或正位模型、在条件基因敲除小鼠中的自发性肿瘤模型以及流式细胞术,探索基质金属蛋白酶9(MMP9)在肿瘤进展和免疫逃逸中的生物学功能。利用单细胞RNA测序和蛋白质组学来深入了解MMP9的潜在机制。在CTNNB1GOF HCC中,MMP9显著上调。MMP9抑制了CD8+ T细胞的浸润和细胞毒性,这对于CTNNB1GOF引发抑制性肿瘤免疫微环境(TIME)和抗PD-1抗性至关重要。从机制上讲,CTNNB1GOF下调了sirtuin 2(SIRT2),导致促进β-连环蛋白/赖氨酸去甲基酶4D(KDM4D)复合物形成,促进MMP9的转录激活。HCC分泌的MMP9介导了CD8+ T细胞中slingshot蛋白磷酸酶1(SSH1)的脱落,导致抑制C-X-C motif趋化因子受体3(CXCR3)介导的G蛋白偶联受体内的信号传导。此外,MMP9阻断重塑了TIME,并增强了在HCC中抗PD-1疗法的敏感性。CTNNB1GOF通过激活MMP9的分泌引起抑制性TIME。针对MMP9重塑TIME并增强CTNNB1GOF HCC中抗PD-1的疗效。
该研究于2023年12月发表在《Gut》,IF:24.5。
技术路线:

结果:
1、MMP9 在 CTNNB1GOF HCC 中表达上调
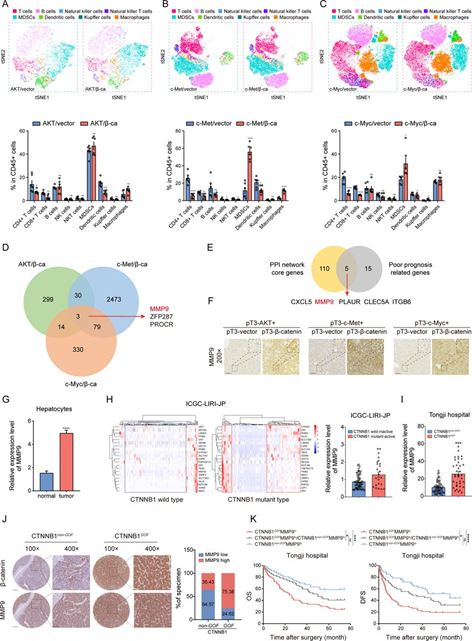
为了识别由CTNNB1GOF驱动的激活基因,我们通过流体动力学尾静脉注射(HTVi)激活的AKT/β-连环蛋白、c-Met/β-连环蛋白和c-Myc/β-连环蛋白的三种HCC模型构建了具有CTNNB1GOF背景的三种类型。ΔN90-β-连环蛋白质通过HTVi与另一个致癌基因质结合,促使肝脏中的肿瘤发生。然后,使用流式细胞仪(FCM)和t-分布式随机邻域嵌入(t-SNE)分析对肝脏免疫微环境进行了分析。与先前的报告一致,在CTNNB1GOF HCC中,巨噬细胞和骨髓源性抑制性细胞的浸润增加,而DCs、CD8+和CD4+ T细胞减少(图1A–C)。与此同时,CD8+ Granzyme B+ T细胞减少,表明CTLs的细胞毒性受损。由于肿瘤负担较高,实验组的总生存期(OS)缩短。随后,对三对模型进行了RNA-seq。韦恩图显示了3个上调基因(MMP9、PROCR、ZFP287)的重叠(折叠变化(FC)>1.0,p<0.05)(图1D)。为了识别与免疫调节和不良预后相关的核心基因,我们通过在癌症基因组学数据库(TCGA)数据集中使用蛋白质-蛋白质相互作用网络(PPI网络)和单变量COX回归对ImmuneScore和StromalScore筛选的差异表达基因(DEGs)进行了分析。结果,确定了五个基因(CXCL5、MMP9、PLAUR、CLEC5A、ITGB6)(图1E)。这两个基因集的交集确定MMP9是潜在的CTNNB1GOF驱动基因,可能在HCC中重塑TIME。然后,验证了MMP9在CTNNB1GOF肿瘤中高表达(图1F)。
MMP9在多个基因表达均衡数据库中都被证实在HCC中上调。此外,根据Lu的单细胞测序数据,肝细胞中MMP9在HCC进展过程中呈最显著升高(图1G)。此外,我们使用国际癌症基因组学联盟数据集中的21基因特征调查了CTNNB1的活化状态。结果显示,在CTNNB1突变的活跃状态下,MMP9上调(图1H)。MMP9在CTNNB1GOF HCC中的上调也在同济队列中观察到(图1I和J)。与此同时,CTNNB1GOFMMP9高表达的肿瘤与更差的生存和较低的无病生存率相关(图1K)。这些结果表明MMP9在CTNNB1GOF HCC中上调,并与不良预后相关。
2、MMP9通过诱导抑制性的免疫微环境(TIME)促进HCC的进展
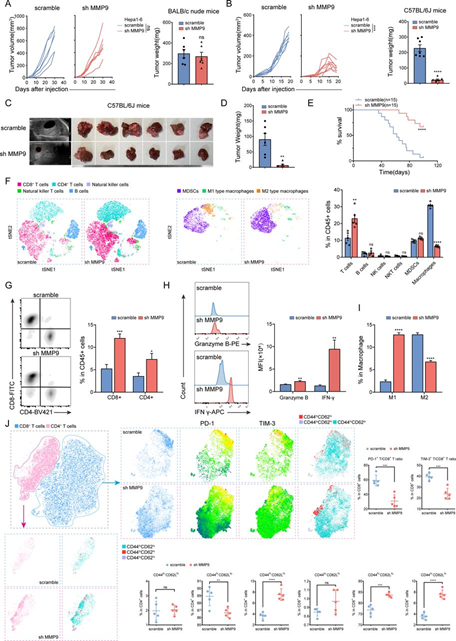
基因集富集分析(GSEA)显示MMP9表达与TCGA-LIHC数据集中的抑制性免疫微环境(TIME)相关。MMP9在体外和BALB/c裸鼠的肿瘤增殖中几乎没有影响(图2A)。然而,MMP9的敲低显著减少了C57BL/6 J小鼠的肿瘤体积和重量(图2B)。对两组小鼠脾脏的流式细胞仪(FCM)分析确认,尽管T淋巴细胞的比例相近,但在BALB/c裸鼠中成熟的CD4+和CD8+ T细胞几乎不存在。
接下来,使用MMP9敲低处理的Hepa1-6细胞构建了原位HCC模型,结果显示明显抑制了肿瘤形成并改善了生存(图2C–E)。FCM分析显示MMP9敲低组中总T淋巴细胞、CD8+和CD4+ T细胞增加,而巨噬细胞减少(图2F和G)。同时,CD8+ T细胞中Granzyme B和干扰素γ(IFN-γ)的增加表明CTLs的细胞毒性增强(图2H)。免疫组化(IHC)和多重免疫荧光(mIF)分析进一步证实了这些结果。尽管巨噬细胞的总数减少,但M2表型向M1表型的转变得以观察(图2I)。随后的分析显示CD8+ T细胞中PD-1和T细胞免疫球蛋白和黏液结合域-3(TIM-3)减少(图2J)。此外,使用CD44和CD62L标记CD8+和CD4+ T细胞的结果表明,在MMP9敲低后,免疫记忆反应水平提高(图2J)。总的来说,这些发现表明MMP9通过诱导抑制性的TIME促进了HCC的进展。
3、MMP9 抑制 CD8+ T 细胞的激活和迁移
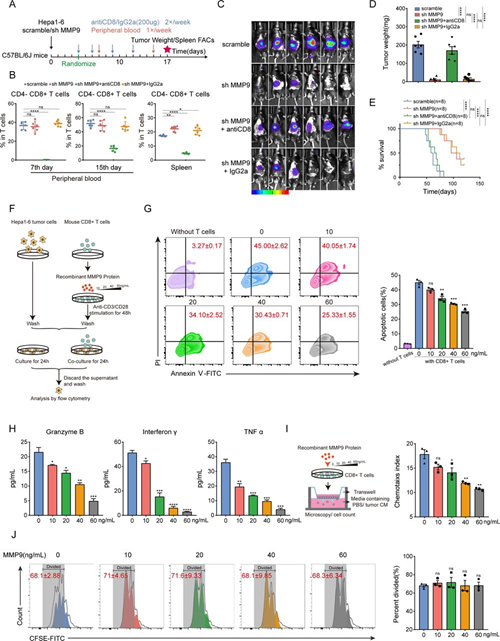
考虑到在MMP9下调后T淋巴细胞和巨噬细胞极化均发生了变化,我们通过消除CD4+ T细胞、CD8+ T细胞或巨噬细胞来进一步确定MMP9调控的特定免疫细胞亚群。分别使用抗CD4中和抗CD8中和抗补体进行原位HCC模型处理(图3A)。通过FCM监测清道夫的效果(图3B)。CD8+ T细胞的消耗剥夺了MMP9下调的抑制肿瘤效果,导致更重的肿瘤负担和缩短的生存期(图3C–E)。相反,CD4+ T细胞和巨噬细胞的消耗无法恢复肿瘤生长。这些结果确认了抑制CD8+ T细胞对于MMP9促进肿瘤进展至关重要。
为了揭示MMP9对CD8+ T细胞激活的影响,进行了体外共培养研究。小鼠脾脏CD8+ T细胞受到重组MMP9蛋白的刺激,并与Hepa1-6细胞共培养24小时(图3F)。CD8+ T细胞的肿瘤毒性在MMP9刺激下呈剂量依赖性减弱(图3G)。CD8+ T细胞在MMP9刺激下的细胞培养上清Elisa测定显示,MMP9抑制了肌酸酶B、干扰素γ和肿瘤坏死因子α(TNF-α)的分泌(图3H)。为了描绘MMP9在CD8+ T细胞迁移中的作用,将肿瘤条件培养基(CM)添加到下层培养皿,并评估CD8+ T细胞向下层培养皿的迁移(图3I)。结果表明MMP9抑制了CD8+ T细胞的趋化作用(图3I)。此外,MMP9不影响CD8+ T细胞的增殖(图3J)。总的来说,这些观察结果表明MMP9阻碍了CD8+ T细胞的激活和迁移,导致了抑制性的免疫微环境。
4、MMP9 阻断可改善 CTNNB1GOF HCC 中的 TIME 并增强抗 PD-1 功效
基于抗PD-1的免疫疗法已广泛用于HCC治疗,并且已报道肿瘤浸润的CD8+ T细胞是抗PD-1疗法的生物标志物。鉴于MMP9对CD8+ T细胞的影响,我们推测MMP9的阻断可能增强抗PD-1疗法的疗效。
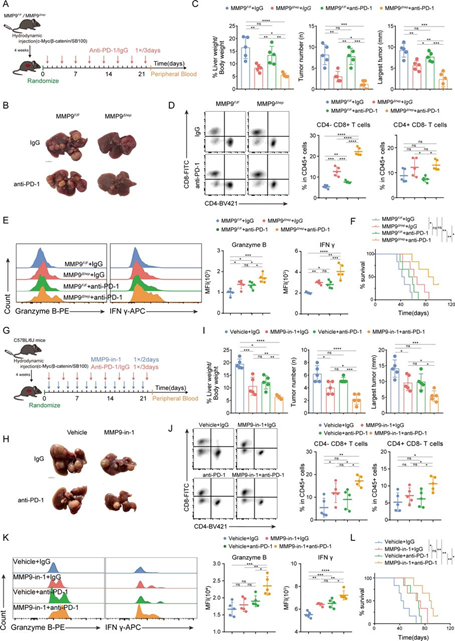
为了评估MMP9阻断对CTNNB1GOF HCC中抗PD-1疗效的影响,我们在MMP9肝细胞条件敲除小鼠(MMP9F/F; MMP9-Alb-cre,以下简称MMP9Δhep)中构建了CTNNB1GOF自发性肿瘤模型。在肿瘤形成后,一部分小鼠接受了抗PD-1抗体治疗(图4A)。通过肝形态学、H&E染色和免疫组织化学(IHC)的验证,所有小鼠均被确定为携带CTNNB1GOF的HCC模型(图4B)。值得注意的是,与其他组相比,接受抗PD-1抗体治疗的MMP9Δhep小鼠显示出显著的肿瘤抑制和生存延长(图4C,F)。流式细胞仪分析显示,与其他组相比,接受抗PD-1治疗的MMP9Δhep小鼠CD8+细胞显著增加(图4D)。CD8+ T细胞中Granzyme B和IFN-γ的增加表明CTLs的细胞毒性增强(图4E)。此外,联合治疗组的多器官未观察到病变,表明MMP9阻断和抗PD1的联合治疗总体上是安全的。血常规和血生化分析显示,联合治疗有效缓解了肿瘤负担引起的肝脏和肾脏损伤。
我们进一步引入了MMP-9-in-1,一种特异的MMP9抑制剂,选择性靶向血浆蛋白饱和度(PEX)。类似地,MMP-9-in-1与抗PD-1联合治疗CTNNB1GOF HCC模型(图4G)。一致地,与MMP-9-in-1或抗PD-1单独治疗相比,联合治疗导致更为显著的肿瘤回归和生存延长(图4H1和L)。流式细胞仪分析显示,联合治疗增强了CD8+ T细胞的浸润和细胞毒性(图4J和K)。联合治疗的安全性也通过相同的方式得到验证。
这些结果表明,MMP9阻断有潜力重塑CTNNB1GOF HCC的免疫微环境,并增强对抗PD-1治疗的敏感性。
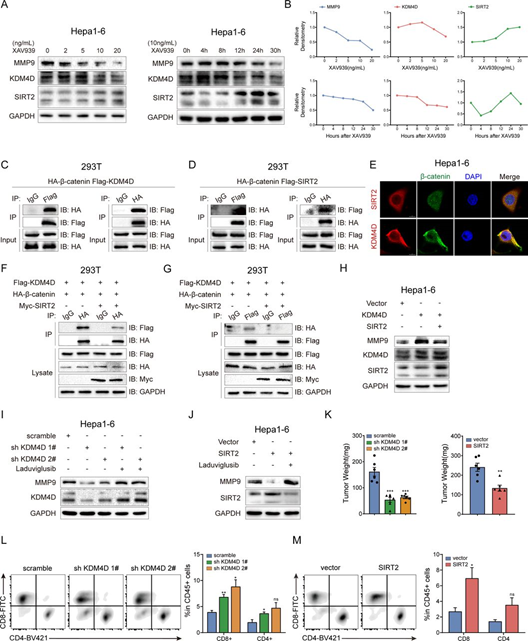
5、β-连环蛋白介导的 SIRT2 抑制通过促进 β-连环蛋白/KDM4D 复合物的形成上调 MMP9
我们进一步研究了β-catenin调控MMP9的精确机制。在组装转录复合物之前,β-catenin与各种蛋白相互作用形成不同的蛋白复合物,执行不同的功能。据报道,KDM4D与β-catenin相互作用,导致MMP9启动子上H3K9me3的去甲基化。SIRT2可以直接与β-catenin相互作用,抑制Wnt信号输出,而抑制Wnt/β-catenin信号增强SIRT2启动子活性。我们发现XAV939通过激活Wnt/β-catenin信号途径促进了KDM4D的表达,并以剂量依赖和时间依赖的方式抑制了SIRT2的表达(图5A,B)。共免疫沉淀(CoIP)结果验证了β-catenin与KDM4D或SIRT2的结合(图5C,D)。免疫荧光证实了在Hepa1-6细胞的细胞质和细胞核中β-catenin与KDM4D或SIRT2的共定位(图5E)。
基于以上观察,我们假设SIRT2和KDM4D可能与β-catenin竞争结合。CoIP结果表明,SIRT2的过表达显著抑制了β-catenin与KDM4D的结合(图5F,G)。此外,在KDM4D过表达的Hepa1-6细胞中,SIRT2的过表达限制了MMP9的上调(图5H)。随后,通过KDM4D敲除或SIRT2过表达诱导的MMP9蛋白的下调在laduviglusib的刺激下被逆转(图5I,J)。此外,KDM4D敲除或SIRT2过表达抑制了MMP9的表达,并导致TIME的激活,从而抑制了肿瘤的形成(图5K–M)。这些结果表明,通过β-catenin抑制SIRT2促进了β-catenin/KDM4D复合物的形成,从而通过上调MMP9促使抑制性TIME的形成。
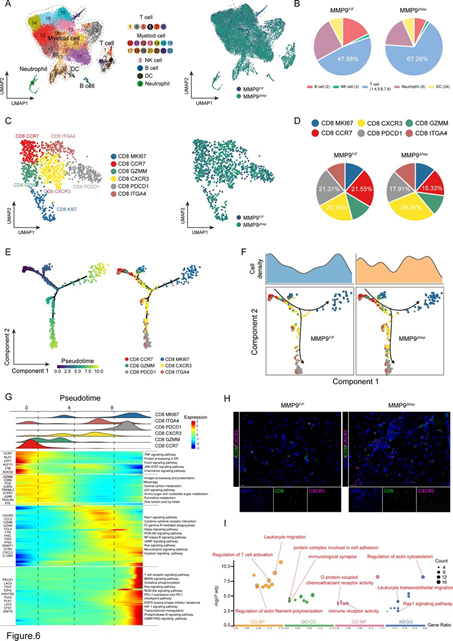
6、MMP9 抑制 CTNNB1GOF HCC 中 CD8+ CXCR3+ T 细胞的浸润
接下来,我们进行了scRNA-seq以探讨MMP9在重塑TIME方面的机制。利用荧光激活细胞分选(FACS)从MMP9F/F和MMP9Δhep小鼠的CTNNB1GOF HCC组织中分离肿瘤CD45+细胞。对总CD45+细胞群体的无监督统一流形逼近和投影(UMAP)分析确定了23个亚群(图6A)。所确定的免疫细胞群体包括T细胞、髓样细胞、NK细胞、B细胞、DCs和中性粒细胞。所有细胞都表达高水平的家庭基因ACTB,而免疫细胞是PTPRC+,AFP,表明我们的数据的准确性。这些细胞亚型在MMP9F/F和MMP9Δhep小鼠中是共享的(图6A)。鉴于髓样细胞的大比例,我们进行了单独的分析以确定髓样和非髓样细胞的比例(图6B)。
在先前建立了MMP9在促进CD8+ T细胞依赖性HCC中的作用后,我们对CD8+ T细胞进行了无监督聚类分析。结果揭示了六个不同的CD8+ T细胞亚型,即CD8 MKI67、CD8 CCR7、CD8 GZMM、CD8 CXCR3、CD8 PDCD1和CD8 ITGA4(图6C)。特别是,MMP9Δhep样本显示了CD8 CXCR3细胞的最显著增加(图6D)。我们观察到CD8 CXCR3细胞的转录谱与已报道的与效应T细胞功能相关的CD8 CX3CR1细胞一致。随后,我们研究了CD8+ T细胞的动态免疫状态。伪时间分析显示,过渡过程始于CD8 CCR7细胞,通过以CD8 GZMM和CD8 CXCR3细胞为特征的中间细胞毒状态,最终导致以CD8 PDCD1和CD8 ITGA4细胞为特征的增殖(CD8 MKI67细胞)或衰竭(图6E)。为了进一步描绘过渡状态,我们分别分析了来自MMP9F/F和MMP9Δhep样本的CD8+ T细胞的轨迹。令人惊讶的是,CD8 CXCR3细胞主要分布在MMP9Δhep样本中(图6F,G)。因此,我们得出结论,MMP9敲除主要影响CD8+ CXCR3+ T细胞的浸润和功能。这一结果得到了mIF染色实验的进一步支持,确认了MMP9Δhep样本中CD8+CXCR3+ T细胞的富集(图6H)。基因本体论(GO)和基因组的京都百科全书(KEGG)通路富集分析表明,CD8+CXCR3+ T细胞特异地富集于G蛋白偶联趋化因子受体活性、调节T细胞激活、白细胞迁移和细胞黏附等方面(图6I)。
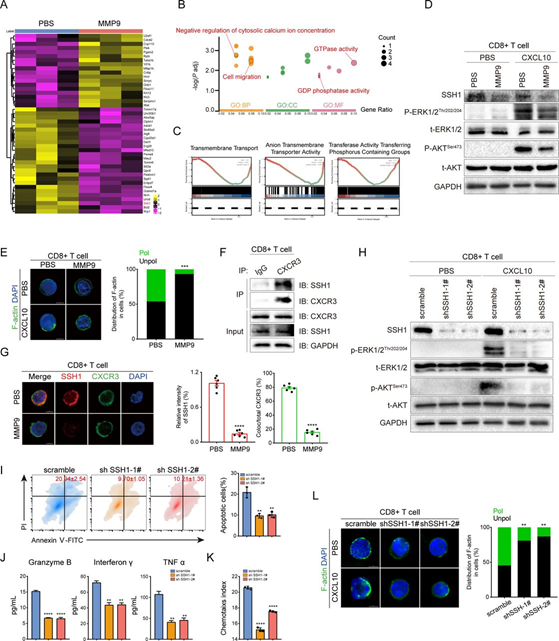
7、MMP9 通过蛋白水解 SSH1 从 CD8+ T 细胞中脱落抑制 CXCR3 介导的 GPCR 细胞内信号传导
我们假设MMP9介导了对CD8+ T细胞表面蛋白的裂解,从而抑制了GPCR介导的CXCR3内部信号传导。蛋白质组学鉴定了在MMP9刺激后CD8+ T细胞中下调的26种蛋白质(图7A),这些蛋白质富集于GTP酶活性、GDP磷酸酶活性、调节细胞质钙离子浓度和细胞迁移等过程(图7B)。GSEA进一步证明MMP9刺激与多种膜转运过程相关联(图7C)。同样,MMP9抑制了CD8+ T细胞中CXCR3介导的ERK和AKT的磷酸化(图7D)。与未受MMP9刺激的T细胞相比,受MMP9刺激的CD8+ T细胞的F-actin分散在细胞外缘,而在T细胞的前缘积聚(图7E)。这些数据表明MMP9抑制了GPCR的CXCR3介导的内部信号传导。此外,在MMP9刺激下,促进定向细胞迁移和T细胞对抗原刺激的SSH1在CD8+ T细胞中显著降低(图7A,D)。 MMP9和SSH1之间观察到的负相关提示了MMP9在SSH1裂解中的潜在作用。
进一步研究了SSH1在CD8+ T细胞中的作用。CoIP和免疫荧光染色表明SSH1可以与CXCR3结合,但在MMP9介导的SSH1裂解时解离(图7F,G)。在CXCL10刺激下,CD8+ T细胞中SSH1的敲除抑制了ERK和AKT的磷酸化刺激(图7H),并阻碍了CD8+ T细胞的激活、迁移和极化(图7I–L)。此外,SSH1的上调消除了MMP9诱导的CD8+ T细胞迁移和激活的抑制作用。总体而言,这些发现表明MMP9通过裂解CD8+ T细胞表面的SSH1来阻碍GPCR的CXCR3介导的内部信号传导。
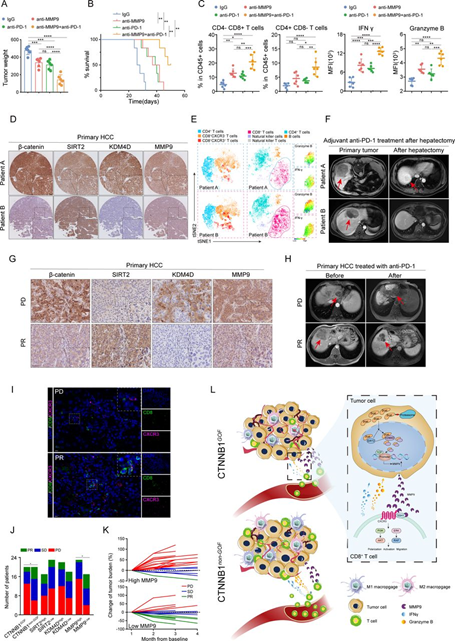
8、MMP9 为 HCC 免疫治疗提供了有价值的治疗靶点和生物标志物
这些研究启发我们提出通过靶向MMP9来开发新的治疗方法。开发了一种新的抗MMP9兔单克隆抗体(mAb),并评估了其在临床实践中的潜在治疗价值。无论是抗PD-1还是抗MMP9单药疗法都在一定程度上抑制了肿瘤生长(图8A),而联合治疗则更显著地抑制了肿瘤生长并延长了生存(图8A和B)。流式细胞仪分析显示联合治疗增强了CD8+ T细胞的浸润和细胞毒性(图8C)。此外,在治疗组中没有观察到明显的毒性,确保了抗MMP9 mAb的安全性。总的来说,这些结果显示了抗MMP9 mAb的治疗价值,抗MMP9和抗PD-1的联合可能是HCC免疫治疗的有前景的策略。
在临床上,我们通过肝切除术或活检从同济医院收集了原发性HCC标本进行进一步的分析。在接受术后抗PD-1辅助治疗的队列中(n=38),免疫组化染色结果显示,MMP9表达与β-连环蛋白和KDM4D呈正相关,而与SIRT2呈负相关(图8D)。tSNE分析显示,MMP9水平较高的患者肿瘤内CD8+CXCR3+T细胞浸润减少,CD8+T细胞细胞毒性减弱(图8E)。此外,MMP9表达水平较高的患者术后复发的可能性较大,与MMP9水平较低的患者相比(图8F)。类似地,对于接受抗PD-1新辅助治疗的患者(n=40),MMP9水平较高导致抑制性TIME和对抗PD-1治疗的响应率较低(图8G–K)。
实验方法:
流式细胞术、tSNE、RNA-seq、scRNA-seq、免疫印迹、免疫共沉淀、RT-PCR、双荧光素酶报告测定、免疫组织化学、多重免疫荧光染色、构建质粒、慢病毒转导、细胞毒性测定、Transwell、F-肌动蛋白染色、串联质量标签 (TMT) 标记的蛋白质组学。
参考文献:
Cai N, Cheng K, Ma Y, et al. Targeting MMP9 in CTNNB1 mutant hepatocellular carcinoma restores CD8+ T cell-mediated antitumour immunity and improves anti-PD-1 efficacy. Gut.