






EGFR激活肌成纤维细胞促进胰腺癌转移
胰腺导管腺癌(Pancreatic Ductal Adenocarcinoma,PDAC)是一种高度侵袭性和难治性疾病,预后很差。癌症相关成纤维细胞(Cancer-associated Fibroblasts,CAFs)是公认的潜在治疗靶点,但对异质性细胞群的了解不足限制了治疗。本研究表明肌成纤维细胞(myofibroblastic CAFs,myCAFs)中转化生长因子β(Transforming Growth Factor Beta,TGF-β)通过双调蛋白介导的自分泌过程诱导EGFR/ERBB2信号的传导。在PDAC类器官衍生培养物和小鼠模型中抑制EGFR/ERBB2信号对不同CAF亚型的影响不同,为其异质性的机制提供了见解。值得注意的是,EGFR激活的myCAFs促进小鼠PDAC转移,揭示了myCAF异质性中的功能意义。最后,对其他癌症数据集的分析表明,这些过程可能在其他恶性肿瘤中起作用这些数据为myCAF异质性提供了功能相关性,并确定了PDAC中预防肿瘤侵袭的候选靶点。
该研究于2023年12月28日发表在《Cancer Cell》,IF:50.3。
技术路线
主要研究结果
1. TGF-β和PDAC类器官条件培养基激活myCAFs中的EGFR/ERBB2信号传导
TGF-β信号传导促进PDAC myCAFs的形成和增殖,但尚不清楚该途径是否在这些细胞中发挥其他功能。因此,作者描述了PDAC CAF前体细胞——胰腺星状细胞(Pancreatic Stellate Cells,PSCs)——TGF-β处理后受体酪氨酸激酶(RTK)的磷酸化。磷酸化表皮生长因子受体(p-EGFR)和磷酸化Erb-B2受体(p-ERBB2)是TGF-β处理后激活的最丰富的RTK,与对照培养基中培养的静止PSCs相比,它们的水平显著增加(图1A和1B)。此外,单细胞RNA测序(scRNA-seq)数据集的分析证实了EGFR和ERBB2在小鼠和人中PDAC CAFs的表达。PSC中TGF-β受体II(TGFBR2)的缺失阻断了TGF-β应答基因的诱导、TGF-β依赖性增生和EGFR的激活。这表明TGF-β通过其同源受体TGFBR2激活EGFR。此外,PSC中EGFR和ERBB2的激活快速且持续抑制TGF-β受体I(TGFBR1)(TGFBR1i)(图1C-E)。由于TGF-β治疗4天后仍观察到持续的EGFR/ERBB2激活,作者在该时间点对TGF-β-处理的PSCs和对照进行RNA-seq。TGF-β处理的PSCs富集了已知的myCAF相关途径,包括细胞外基质(ECM)相关、TGF-β-依赖性LRRC15+CAF和iCAF特征(图1F、1G)。这些结果验证了TGF-β处理的PSCs myCAFs中激活的信号通路。此外,该分析显示TGF-β诱导的myCAFs中胆固醇生成相关特征显著富集(图1G)。此外,TGF-β处理PSCs还诱导了与EGFR激活相关的特征,包括KRAS信号传导、MAPK信号传导和Dusp6(ERK通路的已知靶点)表达增加(图1F和1G)。
在体外和体内,PDAC恶性细胞均表达TGF-β。此外,PDAC类器官条件培养基(CM)可激活PSC中TGF-β通路的下游成员SMAD2,而在CM处理的PSC中,抑制TGF-β信号传导可增强iCAF表型。这些结果表明,PDAC类器官CM治疗PSC可激活myCAF。因此,在PDAC类器官分泌TGF-β基础上,评估了PDAC类器官CM是否激活PSC中的EGFR/ERBB2信号传导。PDAC类器官CM诱导PSC中的EGFR和ERBB2激活,这被EGFR和ERBB2受体双重抑制剂(ERBBi)内拉替尼阻断(图2A)。除了TGF-β,PDAC类器官还表达EGFR/ERBB2配体,这可能有助于促进myCAFF中EGFR/ERBB2的激活。为了更好地表征EGFR/ERBB2激活的CAFs,作者在存在或不存在ERBBi的情况下对CM处理的PSCs进行RNA-seq。通过将TGF-β和CM诱导的基因与EGFR/ERBB2抑制下调的基因交叉,发现了体外myCAF衍生的52个基因的ERBB特征(图2B)。CM处理的PSCs上调了KRAS信号传导和Dusp6表达,这些效应被ERBBi阻断,而没有显著改变TGF-β信号传导激活(图2C、2D)。此外,胆固醇b生成相关特征是myCAF衍生的ERBB特征中最显著丰富的途径之一(图2C和2D)。
总之,这些数据支持PDAC恶性细胞分泌的TGF-β激活小鼠和人PDAC中myCAFs中EGFR信号传导。
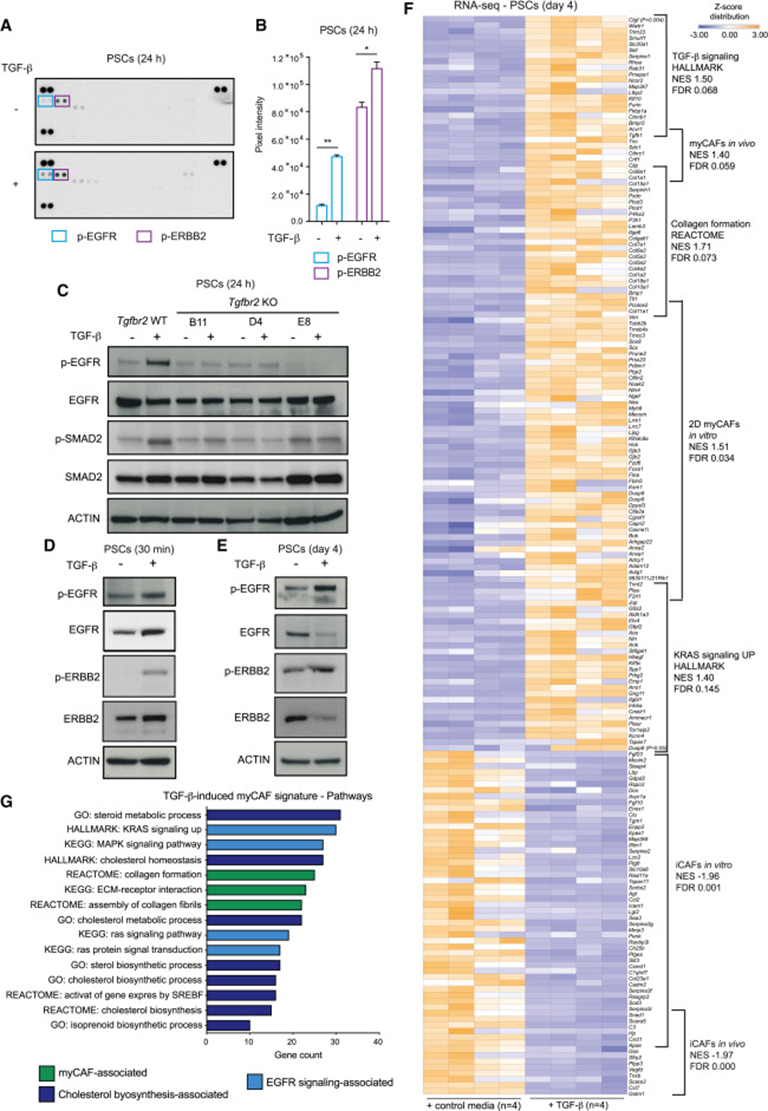
图1:TGF-β激活myCAFs中EGFR/ERBB2信号传导
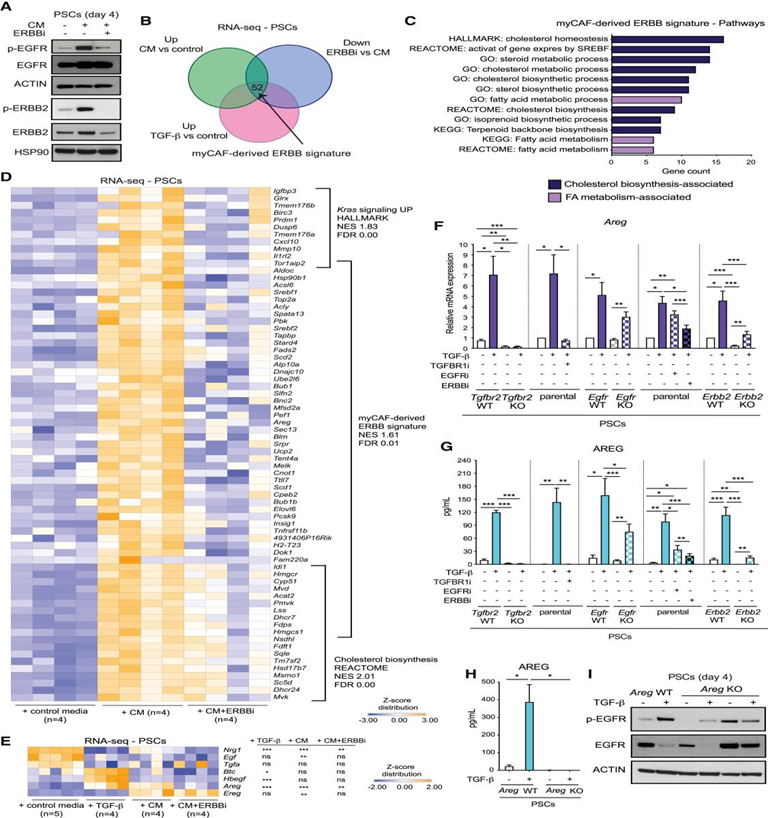
图2:myCAFs中TGF-β诱导的自分泌双调蛋白激活EGFR信号
2.TGF-β诱导的自分泌双调蛋白激活myCAFs中的EGFR信号传导
在TGF-β处理30分钟后,PSCs中EGFR信号的早期激活似乎是由增加的受体介导的(图1D)。为了研究TGF-β诱导的myCAFs中EGFR的激活如何持续,作者在RNA-seq图谱中寻找TGF-β或PDAC类器官CM培养的PSCs,ERBBi存在或不存在时EGFR/ERBB2配体表达。图谱确定了EGFR/ERBB2配体,包括TGF-β显著诱导的双调节蛋白(Areg)和肝素结合EGF-样生长因子(Hbegf)(图2E)。RT-qPCR分析证实了TGF-β-诱导的PSC中Areg和Hbegf的表达,Tgfbr2 KO或TGFBR1抑制剂(TGFBR1i)A83-01治疗阻止Areg的表达(图2F和S2D)。此外,在Egfr或Erbb2缺失或药理抑制后,Areg和Hbegf表达部分缺失,这表明了Areg和Hbegf作为体外TGF-β诱导的myCAFs中EGFR激活的候选介质(图2F)。然而,Areg是TGF-β和PDAC类器官CM治疗显著诱导的EGFR/ERBB2唯一配体(图2E)。此外,PSC中TGF-β上调AREG蛋白,Tgfbr2缺失或TGFBR1药物抑制完全阻断AREG蛋白,但Egfr或Erbb2缺失或抑制并不能阻断AREG蛋白(图2G)。在TGF-β诱导的myCAFs中,直接测试了AREG是否介导EGFR信号的激活,PSCs中删除Areg基因(图2H)。与对照组相比,AREG诱导的EGFR持续激活在AREG KO PSCs中减少(图2I)。
因此,PDAC myCAFs中,自分泌AREG介导EGFR激活下游的TGF-β信号
3. 体外抑制EGFR/ERBB2信号转导耗尽myCAFs
为了进一步了解EGFR/ERBB2激活如何影响myCAFs,首先测量了立即或延迟(72小时)暴露于ERBBi后PSC的增殖,发现增殖显著减少,而凋亡没有增加,这表明EGFR/ERBB2信号传导介导了TGF-β-诱导的myCAFs的增殖(图3A、3B)。此外,体外和体内iCAF的缺氧特征在EGFR/ERBB2抑制后增加,相反的,EGFR/ERBB2抑制下调已知的myCAF相关特征(图3C)。RT-qPCR分析证实了EGFR/ERBB2抑制后iCAF标记的上调(图3D)。当PSCs和PDAC类器官在transwell中共培养时,即使PDAC类器官增殖通过ERBBi治疗而减少(图3E),也观察到这种效果。
为了分析更接近体内情况的模型,建立了PDAC类器官与Egfr WT或Egfr KO PSC的共培养物。然后通过RNA-seq分析流动排序的恶性细胞和PSC群体(图3F和3G)。与对照相比,Egfr耗竭时PSC中大多数下调通路由已知的myCAF相关特征构成(图3H;表S3)。虽然缺氧和体外iCAF特征也被下调,但这可能是由于共培养的恶性细胞的变化,而不是Egfr损失的直接影响。
总而言之,这些数据表明EGFR/ERBB2抑制优先针对体外的myCAFs,且只有myCAFs的亚群被耗尽。此外,这些分析突出显示如何联合EGFR/ERBB2抑制,而不是EGFR封锁单独,可能需要体内有效靶向EGFR激活的myCAFs。
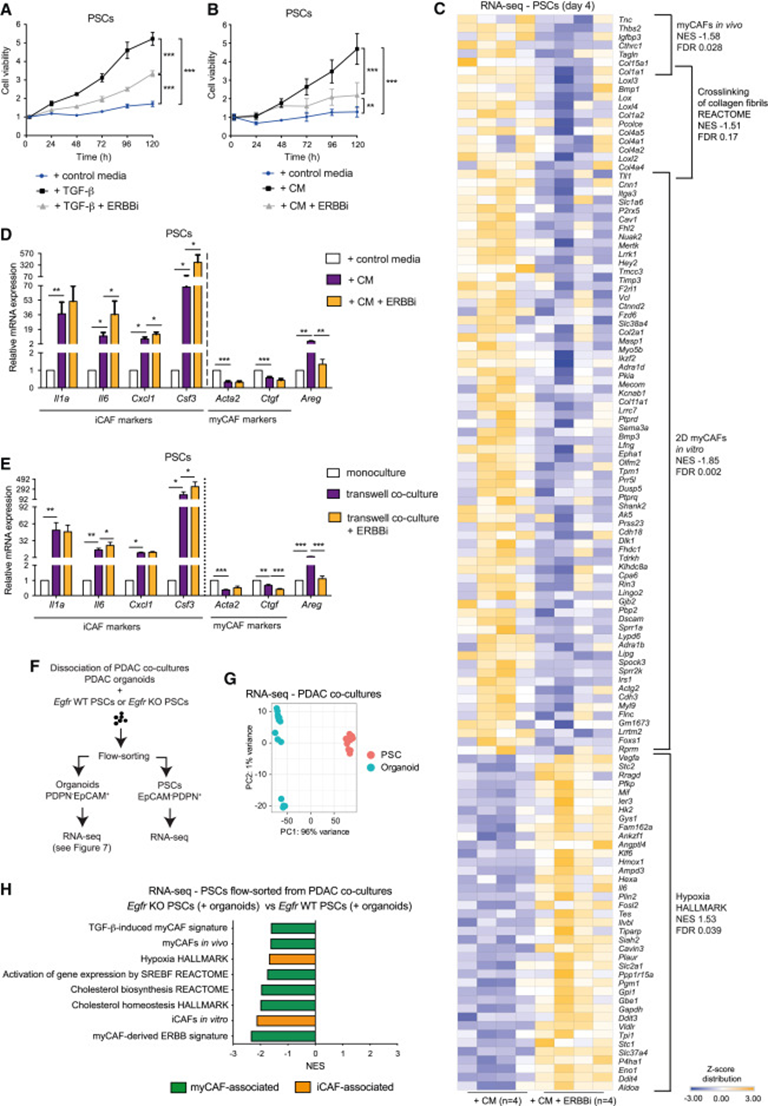
图3:体外抑制EGFR/ERBB2信号转导耗尽myCAFs
4. 抑制EGFR/ERBB2信号优先靶向体内myCAFs
为了确定EGFR/ERBB2信号抑制是否差异影响体内不同的CAF亚型,作者用PDAC类器官建立了原位移植小鼠模型,并用EGFR/ERBB2抑制剂(ERBBi)neratinib治疗荷瘤小鼠2周(图4A)。尽管ERBBi治疗后apCAFs没有显著改变,但myCAFs减少,iCAFs增加,PDAC肿瘤中myCAF/非myCAF的比例显著改变(图4B、4C)。虽然已被证明药物抑制时对CAFs形成途径的相互转换,但这是否与本研究相关仍有待确定。为了进一步研究EGFR/ERBB2抑制对CAFs的影响,作者建立了另外的PDAC类器官来源的原位移植小鼠模型,并在流式分选恶性细胞和成纤维细胞群体进行RNA-seq分析之前,用ERBBi或载体治疗荷瘤小鼠2周(图4A和4D)。与EGFR/ERBB2激活发生在myCAFs中的发现一致,与载体治疗肿瘤的CAFs相比,来自ERBBi治疗肿瘤的CAFs显著下调了Areg表达和TGF-β诱导的myCAF特征,并显著上调了体内iCAF特征(图4E)。此外,先前确定的myCAF亚群如TGF-β依赖性LRRC15+CAFs的特征不受ERBBi治疗的影响,表明EGFR/ERBB2抑制可能只会耗尽myCAFs的一个子集。
为了验证上述发现并进一步研究EGFR/ERBB2抑制后CAF群体的变化,作者对用ERBBi或载体治疗2周的PDAC肿瘤进行了单核RNA测序(snRNA-seq)(图4A、4F、4G)。Dusp6表达的下调证实了多种细胞类型中对该通路的靶向作用,DEGs分析确定上皮细胞和CAFs是EGFR/ERBB2抑制后受影响最大的细胞群体(图4H)。与载体治疗的肿瘤相比,ERBBi治疗的PDAC肿瘤中iCAF丰度和iCAF相关特征丰富,而myCAF丰度和myCAF相关特征下调(图4I-4L)。最后,虽然原发性肿瘤生长和腹水和肝转移的发生率没有显著影响,但EGFR/ERBB2抑制导致膈肌和肺转移的小鼠明显减少(图4M)。
总之,这些数据表明体内PDAC抑制EGFR/ERBB2靶向myCAFs,并表明在myCAFs的一个亚群中的EGFR/ERBB2通路抑制可能会损害PDAC转移形成。
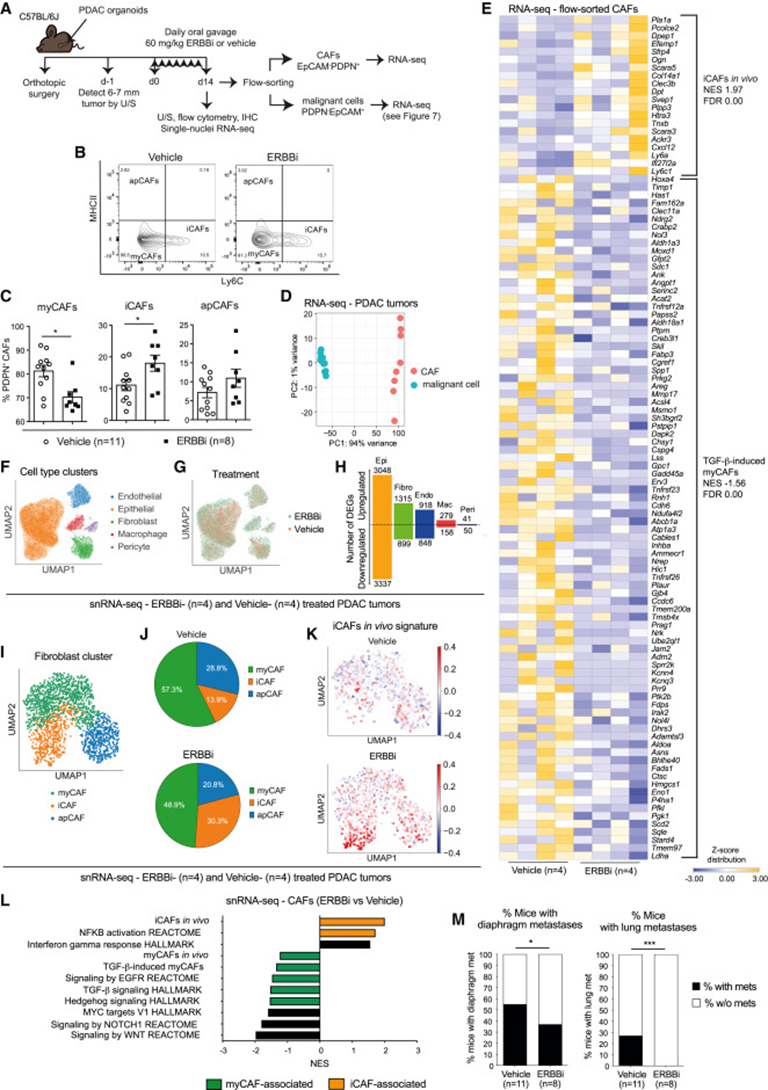
图4:体内抑制EGFR/ERBB2信号优先靶向myCAFs
5. 抑制EGFR/ERBB2信号转导耗尽myCAFs的亚群
虽然数据表明EGFR激活的myCAFs在PDAC中具有潜在的转移促进作用,但有文献表明,α-平滑肌肌动蛋白(αSMA)阳性或HH激活的肌成纤维细胞和细胞外基质成分(如胶原)抑制PDAC进展。与些先前研究以及TGF-β3或HH信号传导抑制相比,ERBBi治疗并未减少整体胶原沉积或肌成纤维细胞标志物αSMA的水平(图5A-5C)。因此,作者评估了EGFR/ERBB2抑制是否仅针对PDAC肿瘤中myCAFs的一个亚群。作者之前研究发现Thy1(编码CD90)在myCAFs中高度表达。为了调查ERBBi治疗对myCAFs子集的潜在差异影响,作者评估了CD90-或CD90+myCAFs(如CD31-CD45-EpCAM-PDPN+Ly6C-MHCII-)丰度的变化。发消息,EGFR/ERBB2抑制对myCAFs的消耗仅限于CD90−myCAFs,在CD90+myCAFs中有适度增加(图5D)。
为了更好地表征CD90- 和CD90+myCAF群体,建立了PDAC的原位嫁接类器官衍生小鼠模型,并在RNA-seq之前对两个myCAF群体进行了流动排序(图5E、5F)。RNA-seq分析证实了两个myCAF群体的流动排序,显示与CD90+ myCAFs相比,CD90-myCAFs中Thy1表达显著下调(图5G)。与CD90+ myCAFs相比,CD90- myCAFs具有显着更高水平的Areg和Dusp6,这表明EGFR信号在CD90- myCAF亚群中更高(图5G)。这些数据为EGFR/ERBB2抑制优先靶向CD90- myCAFs提供了解释。此外,与CD90- myCAFs相比,CD90+ myCAFs具有明显更高的Acta2和Col1a1表达,并且ECM相关特征富集(图5H和5I)。因此,上述数据表明,由于靶向产生较少ECM的CD90-myCAF亚群,EGFR/ERBB2抑制后胶原沉积和αSMA水平没有改变。此外,CD90-myCAFs显著富集胆固醇生物合成相关特征,这些特征在体外TGF-β诱导的myCAF和myCAF-衍生的ERBB特征中上调(图5I、1G和2C)。此外,CD90- myCAFs显示出已知体内iCAF和myCAF特征的显著下调,证实了它们与CD90+myCAFs或其他先前描述的myCAF,包括TGF-β依赖性LRRC15+myCAFs的表型差异(图5I)。最后,除了Areg,还发现了许多在CD90−和CD90+myCAFs中差异表达的其他分泌蛋白,它们可能介导这些CAF群体的不同功能。具体来说,CD90+myCAFs富含胶原蛋白,已被证明在PDAC中起到肿瘤抑制作用,而CD90-myCAFs上调了编码分泌蛋白的基因,包括Spp1和Sema3e,这些蛋白已被证明促进转移(图5J)。
总的来说,这些数据提供了对myCAF异质性的见解,并确定了依赖于EGFR/ERBB2信号激活并可能影响PDAC进展的myCAF亚群。
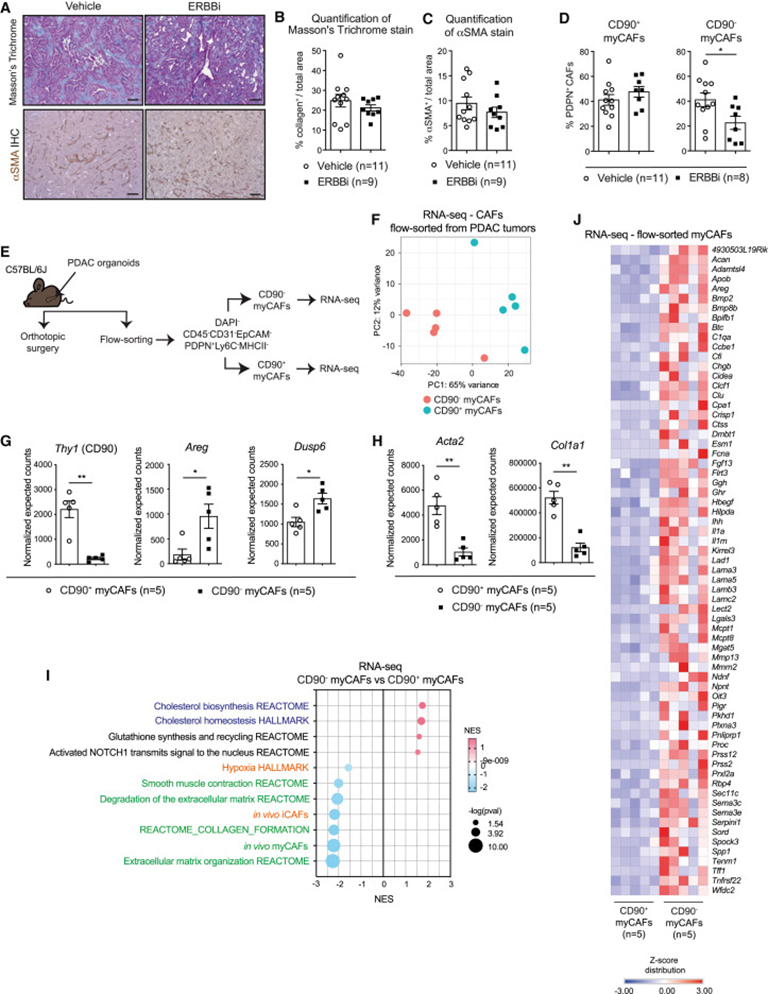
图5:抑制EGFR/ERBB2信号转导耗尽myCAFs亚群
6. EGFR激活的myCAFs促进PDAC转移
在小鼠模型中的ERBBi研究表明,myCAFs中的EGFR/ERBB2抑制可能会损害PDAC转移的形成(图4M)。然而,已有研究报道过PDAC肿瘤中恶性细胞的EGFR信号传导,snRNA-seq分析确定上皮细胞是ERBBi治疗后PDAC肿瘤中受影响最大的细胞类型(图4H)。因此,为了研究EGFR激活的myCAFs在PDAC进展中的作用,建立了PDAC类器官单独或与Egfr WT(如Rosa26 KO)或Egfr KO PSCs共同注射的原位移植小鼠模型(图6A)。通过IHC检测与SV40大T抗原永生的共移植PSC,证实EGFR信号在促进CAF增殖。作者评估了CAFs中Egfr缺失后的改变是否与EGFR/ERBB2抑制后观察到的一致,与PDAC+Egfr WT PSC肿瘤相比,PDAC+Egfr KO PSC肿瘤含有更少的myCAFs,更多的iCAFs(图6B和图6C)。值得注意的是,来自PDAC+Egfr WT PSC的肿瘤明显大于单独来自PDAC的肿瘤(图6D)。此外,与EGFR/ERBB2抑制后观察到的相似,它们产生的隔膜转移、肺转移和腹水明显多于单独PDAC或PDAC+Egfr KO PSC肿瘤(图6E-6I)。
为了研究CAFs中的AREG阻断是否会重现EGFR信号完全消融的影响,建立了PDAC类器官单独或与Areg WT(即Rosa26 KO)或Areg KO PSC(图6J和S6I)共同注射的原位移植小鼠模型。与PDAC+Areg WT PSC肿瘤相比,PDAC+Areg KO PSC肿瘤的巨噬细胞明显减少(图S6J和S6K)。这些发现与体外分析一致,显示Egfr缺失下调但不完全确实PSC中Areg的表达(图2F和2G),并表明myCAF产生的AREG在成纤维细胞-巨噬细胞串扰中的作用。此外,PDAC+Areg KO PSC肿瘤在整体CAF丰度或iCAF/myCAF比例方面与PDAC相比没有显著差异,这些结果表明Areg缺失损害但不完全抑制EGFR信号激活,和myCAF形成(图2I)。PDAC+Areg WT PSC肿瘤明显大于单独来自PDAC或PDAC+Areg KO PSC的肿瘤(图6K)。这些发现也符合体外分析,显示Egfr缺失并不完全降低PSC中Areg的表达,并表明myCAF产生的AREG在局部起作用以促进原发PDAC生长。最后,PSC中的Areg缺失损害了膈肌转移、肝转移和腹水的形成,但对肺转移没有影响,进一步突出了CAF介导的PDAC转移过程的复杂性(图6L-6N)。
总之,这些数据确定了以前未被认识到的myCAFs的功能复杂性,表明EGFR激活的myCAFs促进PDAC的转移。此外,这些结果表明,为了更有效地损害其促转移作用,需要完全消融CAFs中的EGFR信号激活,而不是单独阻断AREG。
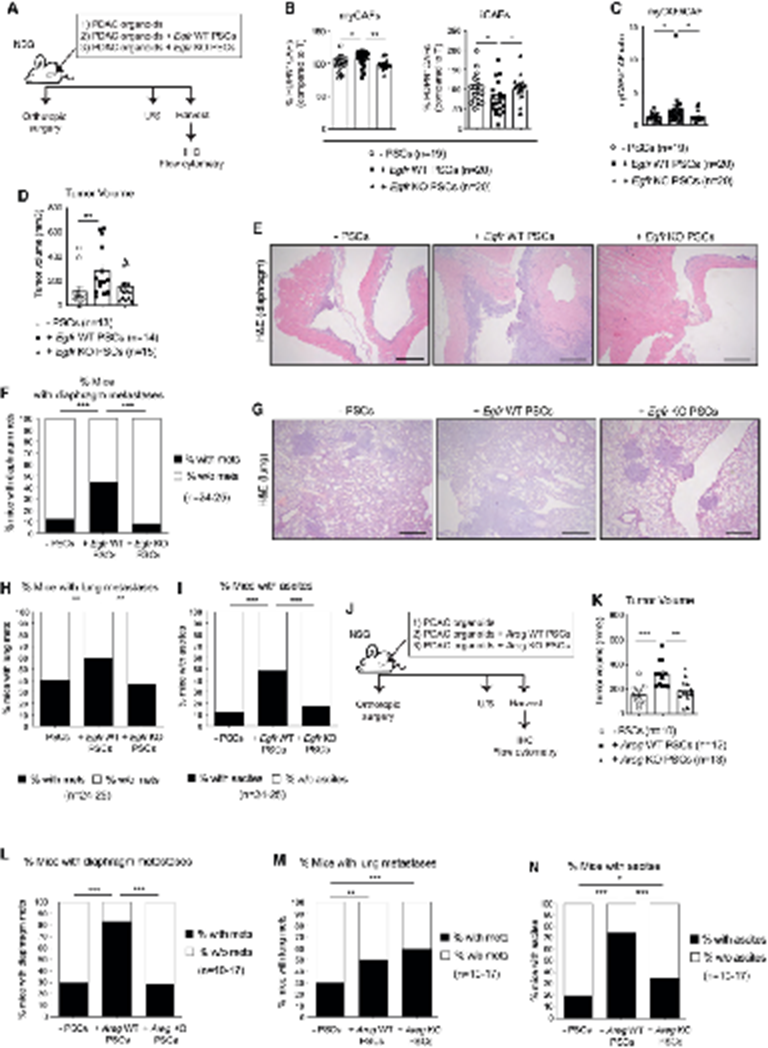
图6:EGFR激活myCAFs促进PDAC转移
7. EGFR激活的myCAFs促进PDAC恶性细胞的转移潜能
为了研究EGFR激活的CAFs促进PDAC转移的潜在机制,对与Egfr WT或Egfr KO PSCs共培养的PDAC类器官中流式分选的RNA-seq进行分析(图3F和3G)。结果看出与Egfr缺失的PSCs培养的类器官中,参与转移形成的通路显著下调,包括EMT转换和缺氧基因特征(图7A)。为了调查这些变化是否也在体内观察到,建立了PDAC类器官与Egfr WT(即Rosa26 KO)或Egfr KO PSCs共注射的原位移植小鼠模型,并对流式分选的恶性细胞进行RNA-seq(图7B和7C)。与PDAC+Egfr WT PSC肿瘤流式分选的恶性细胞相比,DAC+Egfr KO PSC分选出的恶性细胞显示EMT特征下调(图7D)。还分析了体内EGFR/ERBB2抑制后恶性细胞转录组的变化,发现EGFR激活的myCAFs耗尽。ERBBi-或载体治疗2周后,PDAC肿瘤通过流式分选恶性细胞的RNA-seq证实了ERBBi治疗后转移相关通路下调,包括EMT和缺氧基因特征(图4A、4D和7E)。这些结果也通过ERBBi-或载体治疗2周的肿瘤中PDAC恶性细胞的snRNA-seq分析得到证实,该分析显示,与载体治疗的肿瘤相比,ERBBi治疗的肿瘤上皮细胞中EMT和缺氧基因特征显著下调(图4A、4F-4H)。
总之,这些数据支持EGFR激活myCAFs通过增强PDAC恶性细胞的转移潜力促进PDAC转移形成(图7G)。
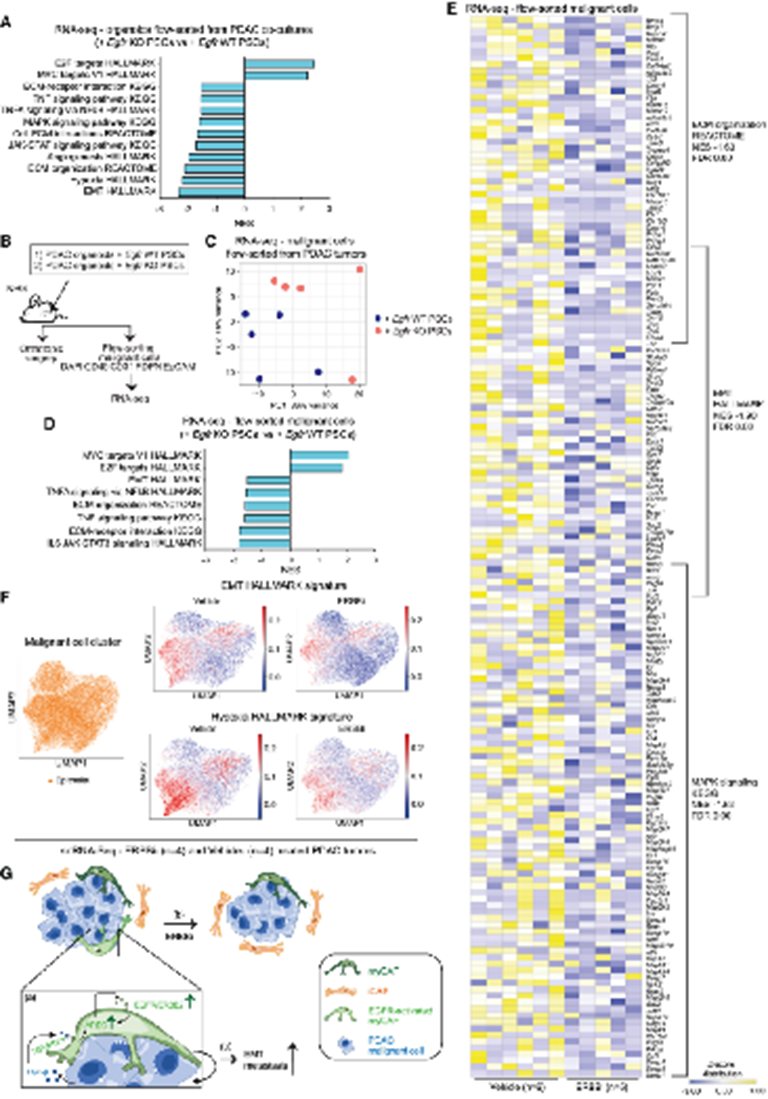
图7:EGFR激活myCAFs促进PDAC恶性细胞的转移潜能
结论
通过互补的体外和体内分析,作者揭示了EGFR激活在PDAC CAFs中以前未知的作用。数据显示TGF-β诱导PDAC myCAFs中的AREG表达,引发自分泌EGFR/ERBB2反应。该网络似乎微调了CAF细胞状态的平衡,相对于iCAF表型,更有利于myCAF。因此,EGFR/ERBB2抑制优先针对myCAFs而不是iCAFs。此外,体内这种效应似乎仅限于EGFR信号激活更高的CD90-myCAFs亚群。最后证明了EGFR激活的myCAFs促进小鼠PDAC转移,从而揭示了双向恶性细胞-成纤维细胞串扰调节PDAC myCAF分子和功能异质性并驱动转移的机制。
实验方法
细胞培养和处理,小鼠实验,原位移植小鼠模型,蛋白质印迹分析,ELISA检测,扩散分析,免疫组织化学和组织学分析,RNA原位杂交分析,流式细胞术分析,细胞共培养,流式分选,逆转录定量聚合酶链反应分析,RNA测序和单细胞RNA测序分析,基因集变异分析,单核RNA测序,降维和聚类,差异基因表达(DEG)分析,基因集富集分析
参考文献
Mucciolo Gianluca, Araos Henríquez Joaquín, Jihad Muntadher, et al. EGFR-activated myofibroblasts promote metastasis of pancreatic cancer.[J] .Cancer Cell, 2023, undefined: undefined.