






scRNA-seq和scATAC-seq联合分析揭示ccRCC异质性和调控动态
透明细胞肾细胞癌(ccRCC)经常具有高度的肿瘤异质性。在单细胞水平上阐明ccRCC的染色质图谱可以加深对该疾病的功能状态和调控动态的理解。在这里,作者对19个ccRCC样本进行了单细胞RNA测序(scRNA-seq)和单细胞转座酶可及染色质测序(scATAC-seq),并利用全外显子组测序来了解个体间的异质性。构建了ccRCC的单细胞转录组和染色质可及性图谱,以揭示ccRCC中不同肿瘤细胞亚型的调控特征。研究发现了两个促进ccRCC侵袭和迁移的长非编码RNA(RP11-661C8.2和CTB-164N12.1),并通过体外实验进行了验证。综上所述,本研究全面描述了ccRCC的基因表达和DNA调控图谱,可为ccRCC的生物学和治疗提供新的见解。本文于2023年3月发表在《Cancer Research》IF:11.2期刊上。
技术路线:

主要实验内容:
1、单细胞多组学研究描绘了人类ccRCC转录组景观
考虑到先前在ccRCC进行的单细胞转录组研究数量,作者希望通过单细胞多组学方案(图1A)进一步发现表观遗传调控机制。从19名患有局部ccRCC的患者中收集了新鲜的手术切除物制备单细胞悬液,一部分用于scRNA-seq,而其余部分则用于scATAC-seq(图1A)。额外的组织或单细胞悬液用于大规模ATAC-seq或WES,如图1A所示。经过Seurat质控后,从ccRCC中捕获到了102,723条高质量的单细胞转录组信息。为减轻批次效应,使用Harmony跨样本汇总细胞并聚类。基于先前研究中标记基因的表达和细胞注释,将ccRCC分类为22个独立的细胞类型,包括四种肿瘤细胞亚型(ccRCC 1、ccRCC 2、ccRCC 3和ccRCC 4)、四种内皮细胞亚型(Endo cells 1、Endo cells 2、Endo cells 3和Endo cells 4)等(图1B和C)。在ccRCC中,发现T细胞(30,339,29.53%)最多,其次是肿瘤细胞(19,819,19.29%;图1D)。由于ccRCC的肿瘤异质性,对来自19名不同个体的肿瘤样本进行了scRNA-seq。发现大多数细胞类型都来自每个样本,而肿瘤细胞和内皮细胞之间的个体差异更为显著(图1E)。例如,肿瘤细胞(ccRCC 4)仅来源于患者RCC84,而Endo cells 4仅来自三名个体(图1E)。与先前研究一致,免疫细胞的广泛浸润,特别是T细胞和TAMs,表征了ccRCC的免疫微环境(图1B和D)。
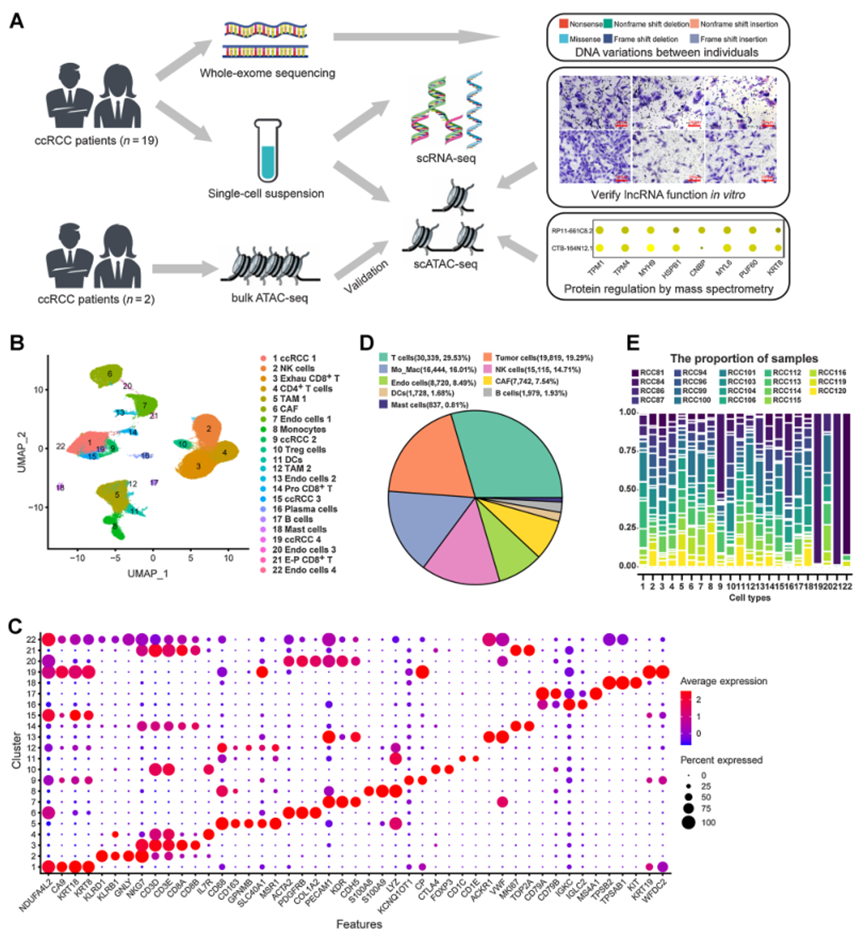
图1 ccRCC样本的单细胞转录组图谱总览
2、通过scRNA-seq对肿瘤细胞的分子亚型进行表征
考虑到ccRCC的遗传倾向,对所有ccRCC样本进行了全外显子测序以了解个体之间的DNA变异。发现这些常见突变基因的变异与先前研究中的相似(图2A)。VHL是最常见的突变基因,其突变率在本研究中为84.2%(图2A)。有趣的是,发现CA9(15.8%)、KRT14(15.8%)和CD24(47.3%)的突变率也较高,这些突变是错义突变(图2A)。将肿瘤细胞分为VHL、CA9、KRT14和CD24突变或非突变组,并比较它们的基因表达差异(图2B–E)。发现突变和非突变的肿瘤细胞基因表达非常相似,皮尔逊相关系数大于0.98(图2B–E)。因此,这也表明在ccRCC中的单一突变基因并没有引起肿瘤细胞所有基因表达的巨大变化。
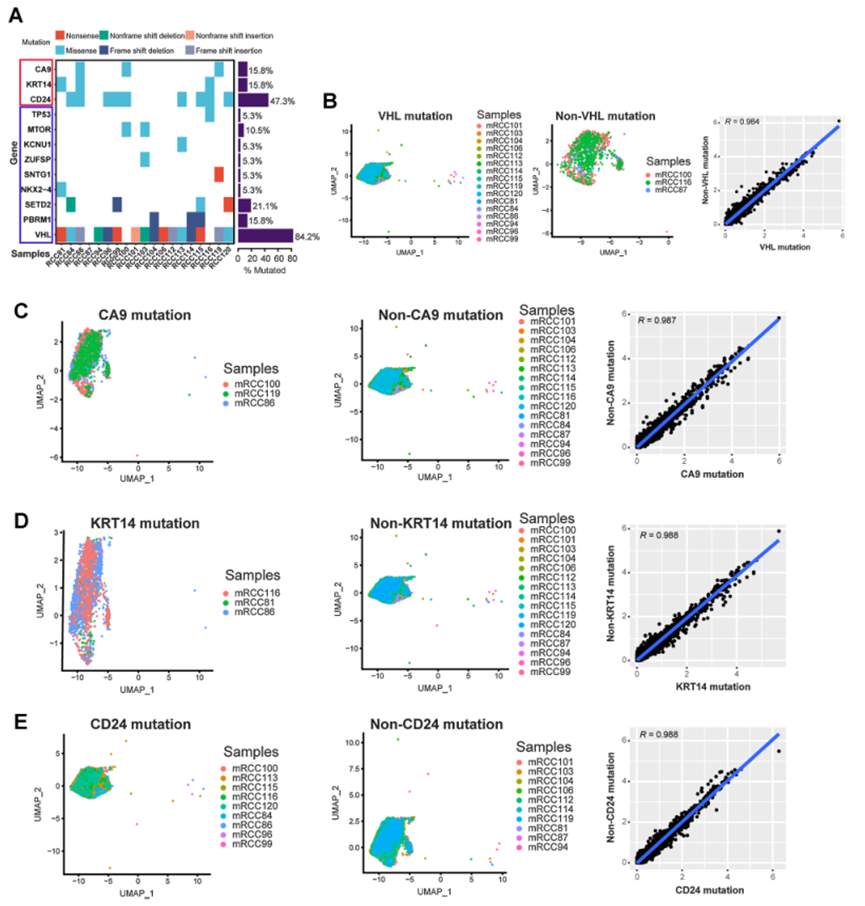
图2 scRNA-seq揭示了ccRCC中基因突变与基因表达的关系
根据基因表达特征,肿瘤细胞通过scRNA-seq(图3A)无偏地聚类成四个细胞亚型(ccRCC 1、ccRCC 2、ccRCC 3和ccRCC 4)。除ccRCC 4外,其余三个肿瘤细胞亚型都来自每个患者(图3B)。尽管存在个体差异,但在肿瘤细胞中有对细胞亚群分类的良好代表性。最初,为客观地揭示肿瘤细胞的特定基因表达,作者将其与邻近正常上皮细胞进行比较,这些细胞来源于作者先前的研究。不同肿瘤细胞亚型中存在基因表达的显著差异。作者发现ccRCC 1高表达CA9和NDUF4AL2,而ccRCC 2高表达KCNQ1OT1和CP(图3C)。ccRCC 3高表达NDUF4AL2和CYB5A,而ccRCC 4表达肿瘤细胞中大多数高表达的基因(图3C)。CA9和NDUF4AL2在先前的研究中已被确认为ccRCC的标记基因。CP在先前的单细胞研究中被确认为ccRCC的特异性标记物,这在本研究中从更大的ccRCC样本中得到了证实。KCNQ1OT1是一个致癌lncRNA,在先前的研究中已有报道。然而,这是通过scRNA-seq首次发现KCNQ1OT1在ccRCC的特定细胞亚型中高表达。
根据scRNA-seq数据,进行拷贝数变异(CNV)分析,发现这些肿瘤细胞亚型存在不同的CNV特征(图3D)。作者发现大多数肿瘤细胞的特征是染色体3(chr3)上的缺失和染色体5(chr5)上的增益,与TCGA对ccRCC的项目结果一致。这个结果在ccRCC 1中更为集中,而在ccRCC 4中不明显(图3D)。这可能是由于肿瘤细胞的克隆或亚克隆导致的。
拥有ccRCC的亚型信息后,作者试图预测这四种肿瘤细胞类型的预后。先计算每个肿瘤细胞类型的差异表达基因(DEGs)。选择每个细胞亚型的前100个DEGs,并基于在TCGA数据库上对ccRCC的生存分析结果确定预后。发现ccRCC 1、ccRCC 2和ccRCC 3中的大多数DEGs表明积极的生存状况,而ccRCC 4中的更多DEGs则表明不良预后,与前三个细胞亚型相比显著(图3E)。
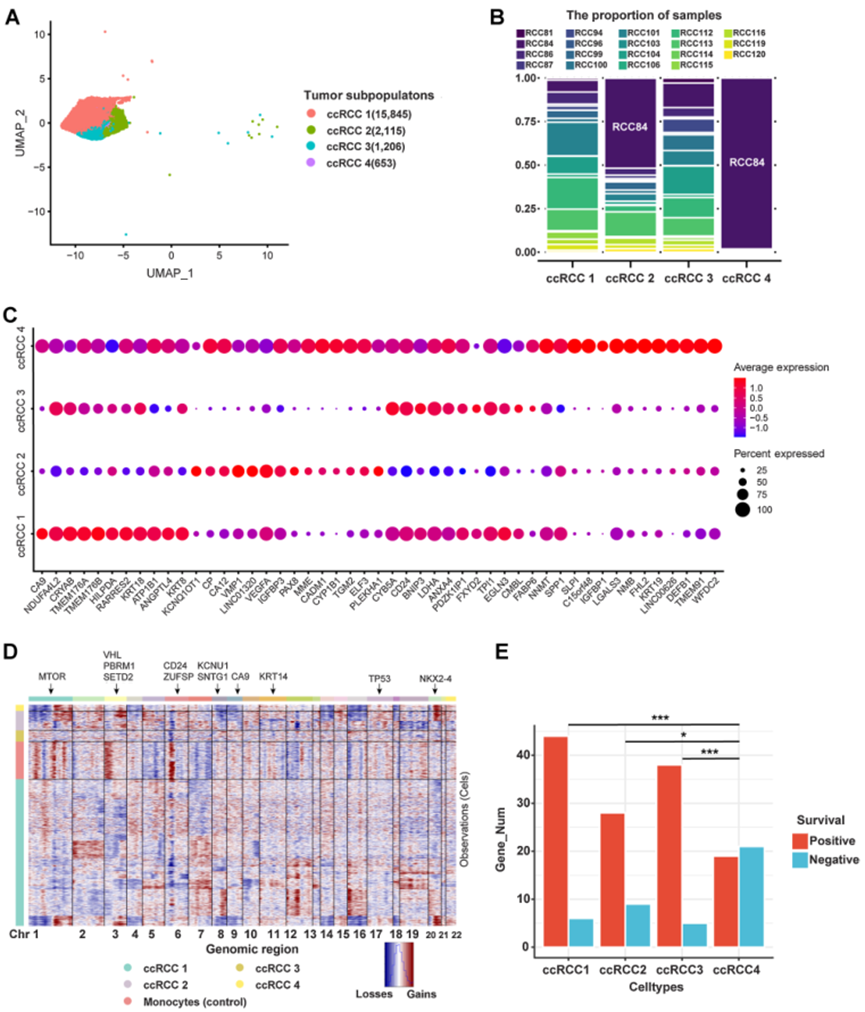
图3 人ccRCC肿瘤细胞的异质性
3、构建ccRCC的单细胞染色质可及性景观
在前一项研究中,已经构建了RCC中免疫细胞动态染色质景观的单细胞图谱,为理解ccRCC中免疫细胞的功能状态和调控动态提供了丰富的资源。然而,需要构建一个更全面的ccRCC单细胞染色质可及性景观,包括肿瘤细胞在内的大样本,这可揭示肿瘤细胞的调控特征。在这项研究中,作者对19个ccRCC样本进行了scATAC-seq。经过质控后,捕获到了61,693个高质量细胞核和190,916个唯一峰值。将这些细胞分类为29种不同的细胞类型,并确定了每种细胞类型的样本来源(图4A和B)。scATAC-seq的细胞注释计算了肿瘤细胞(CA9和KRT14)、内皮细胞(VWF和CDH5)、CAF(RGS5)、免疫细胞(PTPRC)、CD4+ T细胞(IL7R)、B细胞(SDC1)和NK细胞(KLRD1和KLRB1)的标记基因活性分数(图4C)。随后,对特定细胞类型的峰值进行了分析,然后确定了这些峰值的相应基因区域(图4D)。基于以上工作,定义了ccRCC中的29种不同细胞类型(图4A)。
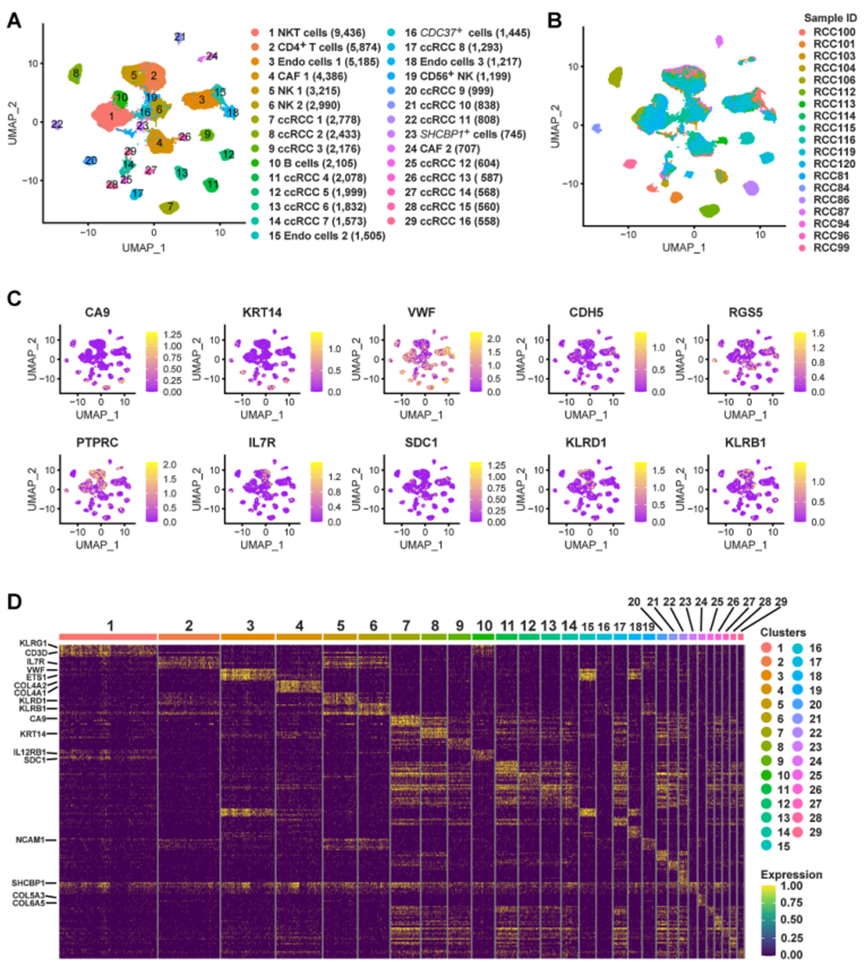
图4 ccRCC的单细胞染色质可及性和外观
利用scATAC-seq在所有细胞类型中确定了共同可及的染色质区域,例如在chr9和chr17中(图5A)。这些共同可及的染色质区域还表明它们的调控过程并非细胞类型特异。因此,通过识别细胞类型特异的共同可及染色质区域,可以发现细胞类型特异的调控过程。此外,scATAC-seq结果显示非肿瘤细胞(如免疫细胞、内皮细胞和CAF)的染色质可及性在个体间是一致的(图5B)。每种细胞类型都具有良好的样本普适性,并且几乎来自所有样本(图5B)。然而,基于染色质可及性,肿瘤细胞可以被分类为16个细胞亚型(图5C)。大多数肿瘤细胞亚型来自单个ccRCC样本(图5C)。类似于先前对基底细胞癌的研究,ccRCC中肿瘤细胞的染色质可及性显示出显著的个体间异质性。
4、scATAC-seq揭示ccRCC肿瘤细胞染色质调控的特点
为研究肿瘤细胞染色质调控的特征,利用scATAC-seq鉴定了16个肿瘤细胞亚型及每个亚型的染色质可及性区域(图5C)。作者发现CA9和KRT14在ccRCC细胞的可及性区域中充当标记基因,它们在每个肿瘤细胞亚型中都保持染色质可及性(图5D和E)。为更好地发现肿瘤细胞亚型之间的异质性,对这16个细胞亚型中的前10个染色质可及性区域进行富集,并将这些区域映射到基因上,发现这些染色质可及性区域既有重叠又有差异,呈现出一种不规则的状态。这也可能表明ccRCC在DNA水平上存在遗传倾向。
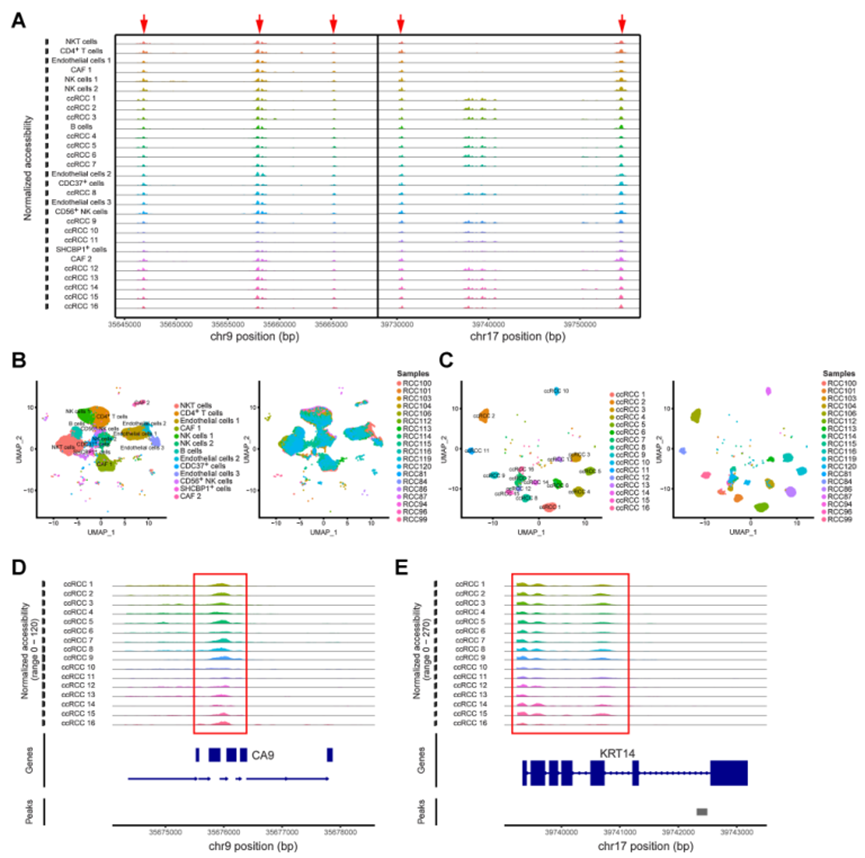
图5 ccRCC单细胞染色质可及性特征
5、scATAC-seq揭示lncRNA在体外促进ccRCC侵袭和迁移
有趣的是,作者通过scATAC-seq发现了四种lncRNA(RP11-661C8.2、CTB-32H22.1、CTB-164N12.1和RP11-267A15.1),这些lncRNA在肿瘤细胞亚型中具有特异的可及性(图6A)。所有这些lncRNA都有一个共同的特征,即位于染色体5上(图6A)。研究证明cis-acting的lncRNA可以激活、抑制或以其他方式调节靶基因的表达。考虑到TCGA和本研究均证实ccRCC具有染色体5上拷贝数增加的特征,作者假设这些lncRNA可能在ccRCC中发挥特定的调控作用。因此,选择了两种lncRNA(RP11-661C8.2和CTB-164N12.1)进行实验,分别利用ASO-5608和ASO-5717敲低 RP11-661C8.2和CTB-164N12.1。随后验证这些lncRNA的生物学功能。
发现经过ASO处理后,ccRCC细胞系的增殖活性显著减少,包括786-O组和Caki-2组(图6B)。CCK-8实验证实ASO处理后ccRCC细胞系的增殖在24、48和72小时内均受到了抑制,与NC组相比(图6C)。NC组的划痕愈合优于ASO处理组,无论是在786-O还是Caki-2中(图6D–F)。此外,Transwell实验结果表明经过ASO处理后,786-O和Caki-2的迁移显著减少(图6G和H)。在ASO处理后,ccRCC细胞系中E-cadherin的表达与NC组相比显著增加(图6I)。这些结果确认RP11-661C8.2和CTB-164N12.1在体外促进ccRCC的侵袭和迁移。
作者通过质谱法鉴定了与RP11-661C8.2和CTB-164N12.1相互作用的蛋白质。根据可信的肽段和蛋白质,富集了与RP11-661C8.2和CTB-164N12.1密切相互作用的蛋白质,并展示最显著的蛋白,如TPM1、TPM4、MYH9、HSPB1、CNBP、MYL6、PUF60和KRT8(图6J)。发现这两种lncRNA结合的蛋白质非常相似,除了CNBP更紧密地与RP11-661C8.2结合(图6J)。随后,将编码这些蛋白质的基因放入scRNA-seq数据中,以估计它们在每个细胞亚型中的表达。有趣的是,这些基因在CAF和内皮细胞3中几乎都高表达(图6J)。一些基因,如TPM1、HSPB1和KRT8,在肿瘤细胞中高表达(图6J)。因此,认为RP11-661C8.2和CTB-164N12.1的调控功能是细胞类型特异的,主要调控CAF、内皮细胞和肿瘤细胞。
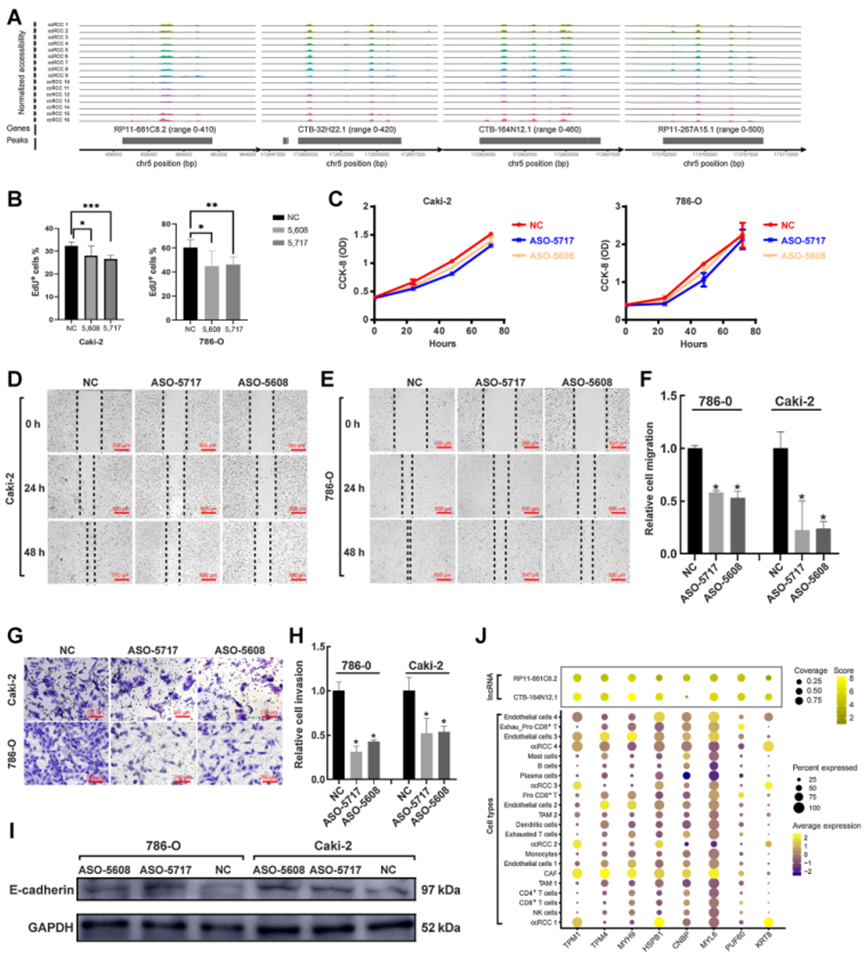
图6 ccRCC肿瘤细胞中特异性lncRNA的发现和验证
6、scATAC-seq揭示ccRCC的转录调控因子
scATAC-seq的另一个优势是发现特定的调控过程,特别是转录因子(TFs)。由于这是单细胞研究,可以将这些TFs特异地定位到细胞类型。作者发现许多具有细胞类型特异性和TF结合基序的TFs。如SPIC和EOMES分别是B细胞和NK细胞中的特异性TFs(图7A)。与先前的研究相比,基序的DNA碱基序列也非常相似(图7A)。作者发现ZEB1充当了一个通用结合肿瘤细胞的TF,而ETV4是被非肿瘤细胞普遍结合的(图7A)。此外,通过scATAC-seq鉴定了内皮细胞(SOX8)、CAFs(EBF2)和T细胞(ETS1)的特异性TFs和基序(图7A)。通过富集每种细胞类型中前10个最显著的TFs,发现这些TFs具有细胞类型特异性,尤其是在肿瘤细胞和免疫细胞之间(图7B)。为预测这些细胞类型特异性TFs的确切结合位置,进行TF足迹分析(图7C)。因此,正如前面提到的,定义scATAC-seq中的细胞类型时需要考虑TFs。
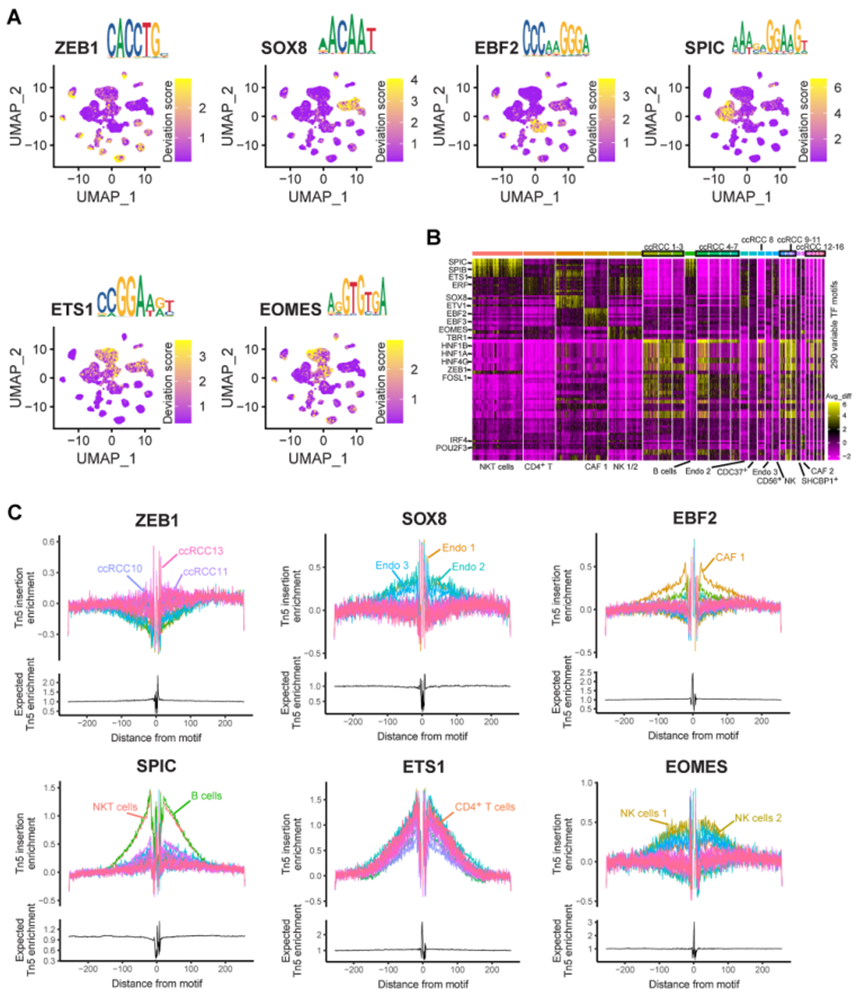
图7 通过scATAC-seq鉴定ccRCC中TFs的特征
7、整合scATAC-seq和scRNA-seq分析揭示肿瘤细胞的调控特征
基因表达的调控是一个非常复杂的过程,受到表观遗传调控、lncRNA、增强子、启动子等因素的影响。在这项研究中,作者通过整合scATAC-seq和scRNA-seq的结果,构建二者的共嵌图发现ccRCC的调控规律(图8A)。尽管通过空间投影反映的scATAC-seq和scRNA-seq数据存在差异,但相同细胞类型之间的分布非常相似(图8A)。
通过整合scATAC-seq(16个亚型)和scRNA-seq(四个亚型)中的所有肿瘤细胞,发现肿瘤细胞亚型中染色质可及性与基因表达之间存在相关性(图8B)。ccRCC 1(scRNA-seq亚型)的基因表达与15个肿瘤细胞类型(scATAC-seq亚型)的染色质可及性相关联(图8C),表明肿瘤细胞调控的复杂性。还发现一对肿瘤细胞类型,即ccRCC 2(scRNA-seq亚型)和ccRCC 11(scATAC-seq亚型),暗示染色质可及性与基因表达之间的对应关系(图8C)。在先前的结果中,ccRCC 2和ccRCC 11主要来自样本RCC84(图2C)。因此,揭示由ccRCC 11的可及染色质区域调控的ccRCC 2的基因表达特征是准确的(图8D)。然而,ccRCC 1(scRNA-seq亚型)的调控特征复杂,包括15个scATAC-seq肿瘤细胞亚型的调控特征(图8E)。尽管如此,在所有15个肿瘤细胞亚型中发现了一些共同的可及染色质区域:“chr11-111906243-111914666”、“chr19-10352456-10354286”、“chr5-178286237-178290844”和“chr7-73829891-73835325”(图8E)。这些发现表明,在研究的ccRCC肿瘤细胞的可及染色质区域存在个体差异,同时也存在共同的调控区域。
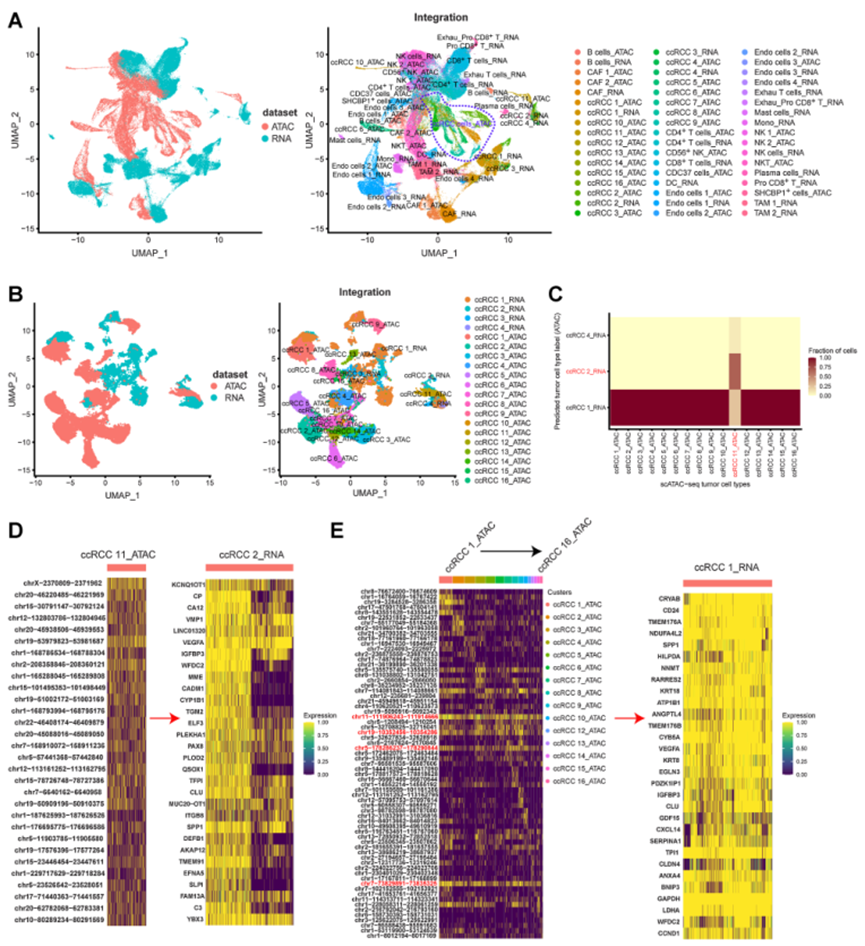
图8 整合scRNA-seq和scATAC-seq分析
实验方法:
临床样本收集,细胞培养,scATAC-seq,scRNA-seq,生物信息学分析, 核/质分离和qRT-PCR,流式细胞术,划痕实验,transwell实验,RNA pull down+质谱,免疫印记
参考文献:
Yu Z, Lv Y, Su C, Lu W, Zhang R, Li J, Guo B, Yan H, Liu D, Yang Z, Mi H, Mo L, Guo Y, Feng W, Xu H, Peng W, Cheng J, Nan A, Mo Z. Integrative Single-Cell Analysis Reveals Transcriptional and Epigenetic Regulatory Features of Clear Cell Renal Cell Carcinoma. Cancer Res. 2023 Mar 2;83(5):700-719. doi: 10.1158/0008-5472.CAN-22-2224. PMID: 36607615; PMCID: PMC9978887.