






TREM2巨噬细胞通过SLC25A53重编程代谢促进心肌梗死心脏修复
心肌梗死(Myculal Infarction,MI)是常见的一种死亡原因,尽管可通过血运重建和药物进行治疗,但MI的死亡率并没有显著下降,对人类健康造成了严重威胁。MI的修复过程中需要单核细胞/巨噬细胞的及时参与以清除死亡的心肌细胞,并保持局部心肌中促炎和抗炎因子之间的平衡。髓样细胞触发受体2(Triggering Receptor Expressed on Myeloid Cells 2,TREM2)参与调节多种细胞反应,包括胞葬、细胞存活和炎症。以往的研究主要集中在TREM2参与慢性疾病,而其在急性疾病(如心肌梗死)中的作用尚不清楚。本研究旨在探讨TREM2在心肌梗死中的作用,并阐明其在心脏修复过程中的功能。具体而言,作者观察到TREM2影响巨噬细胞胞葬糖代谢和抗炎途径,且TREM2在巨噬细胞中的过度表达可以改善心肌梗死后心脏功能的恶化,这是一种有希望的MI治疗方法。总之,本研究揭示了巨噬细胞特异性TREM2在心肌梗死中的新作用,将胞葬与心脏修复过程中的免疫代谢联系起来。
该研究于2024年1月5日发表在《Cell Death & Differentiation》,IF:12.4
技术路线
主要研究结果
1. TREM2+巨噬细胞参与MI早期阶段
作者已确认了表达TREM2的主要细胞类型是巨噬细胞。接下来为了阐明TREM2在MI中的作用,首先检测了心肌梗死后(post-MI)心脏中TREM2的表达,发现TREM2的表达水平显著高于假心梗组,提示TREM2的表达与缺血激发的反应有关(图1A-C)。值得注意的是,post-MI TREM2的表达表现出时间模式。post-MI第1天,TREM2的表达水平开始增加,第3天显著升高,第7天达到峰值,最终恢复到基线(图1A-C)。进一步研究MI不同区域的TREM2表达,发现它主要表达在MI边缘区和缺血区(图1A-C)。.此外,大多数TREM2+细胞为CD68+巨噬细胞,而不是Ly6G+中性粒细胞(图1E-G)。考虑到TREM2+中性粒细胞浸润缺血性心脏组织的转运募集,且TREM2+巨噬细胞在post-MI修复过程中存在时间较长,作者决定将研究重点放在MI中的TREM2+巨噬细胞上。
在WT小鼠中post-MI TREM2+巨噬细胞主要是CCR2+MHCIIlow细胞(图1H,I为第3天和第7天),表明TREM2+巨噬细胞倾向于表现出抗炎表型。此外,对从MI患者获得的外周血单个核细胞(PBMCs)中TREM2的表达水平进行分析,发现MI患者的TREM2的水平升高,且在3-5天观察到水平最高的。重要的是,这种TREM2表达的增加并不归因于循环单核细胞数量的增加,根据人类单核细胞CD14正常表达后(图1J)的实验结果证实。提示MI患者PBMC中TREM2表达的升高可能是由于TREM2表达的诱导,而不是单核细胞群体的变化。
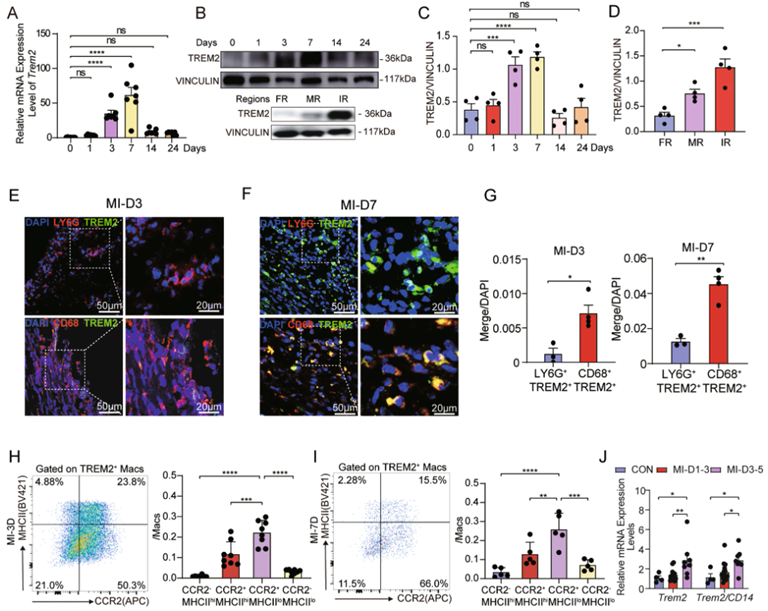
图1:TREM2主要在免疫组织中表达,MI应激诱导TREM2表达
2. 巨噬细胞特异性TREM2缺失加重MI损伤
为了研究TREM2+巨噬细胞对MI的特异性影响,通过LysMCre小鼠与TREM2flox/flox小鼠杂交条件性敲除巨噬细胞中TREM2,并与对照小鼠(TREM2flox/flox)进行了比较。巨噬细胞中TREM2敲除后导致心功能显著下降,如EF和FS减少,以及LVED和LVESV等与post-MI对照组相比增加(图2A,B)。此外,Masson三色染色显示整个心脏的一系列切片纤维化减少,以及Mac-TREM2 KO组梗死区左心室壁厚度显著减少(图2C,D)。接下来,作者对post-MI心脏的死细胞清除、血管生成和纤维化产生进行了组织学评估。与对照组相比,Mac-TREM2 KO小鼠的ACs(凋亡心肌细胞)显著增加(图2E,G)。此外,纤维化标志物,如Vimentin、α-SMA以及增殖标志物Ki67的免疫荧光染色显示Mac-TREM2 KO组纤维化产生较少(图2F,H,I)。这些发现表明Mac-TREM2 KO小鼠坏死细胞清除受损,纤维化产生减少。然而,两组之间的血管生成没有观察到显著差异(图2J)。
此外,对巨噬细胞亚群进行了细胞学评估,发现MI组TREM2敲除小鼠在梗死后7天表现出MHCIIlow巨噬细胞水平显著降低(图2K,L)。这些发现表明TREM2可能调节post-MI巨噬细胞的功能和极化。为了进一步确认在MI中发挥主要作用的巨噬细胞来源,用CCR2趋化因子受体拮抗剂RS504393阻断了循环单核细胞的招募,这不影响常驻巨噬细胞群体。有趣的是,CCR2抑制剂导致对照组EF减少和梗死面积增加,但它不影响Mac-TREM2 KO小鼠的post-MI心脏功能(图2M,N)。这表明在MI中发挥作用的是招募的CCR2+TREM2+巨噬细胞。

图2:巨噬细胞中TREM2的条件敲除对MI小鼠心脏的影响
3. 巨噬细胞中TREM2缺失抑制胞葬
通过生物信息学分析(GSE98563)观察到TREM2巨噬细胞主要与增强的吞噬作用相关(图3A)。此外,吞噬细胞和溶酶体相关基因的表达在TREM2 KO巨噬细胞中明显较低,但在过表达TREM2的巨噬细胞中较高(图3B-D)。为了评估TREM2+巨噬细胞在缺血性心脏病中的胞葬能力,Tdtomato+或ZsGreen+小鼠LAD结扎后从缺血心脏获得的Td+或Zs+标记的ACs与巨噬细胞共培养。体内,Td+或Zs+标记的ACs注射到对照组和Mac-TREM2 KO小鼠的腹腔和心肌中。体外,过表达TREM2的RAW264.7细胞孵育Td+ACs。正如预期的那样,体内和体外实验的结果都支持了TREM2促进巨噬细胞胞葬的假设(图3E-H)。我们进一步评估了心脏内巨噬细胞吞噬心肌细胞的能力。Ter119+CD68+巨噬细胞数量显著减少,普鲁士蓝染色减少,进一步证实post-MI第7天Trem2缺失与巨噬细胞吞噬功能缺陷相关。
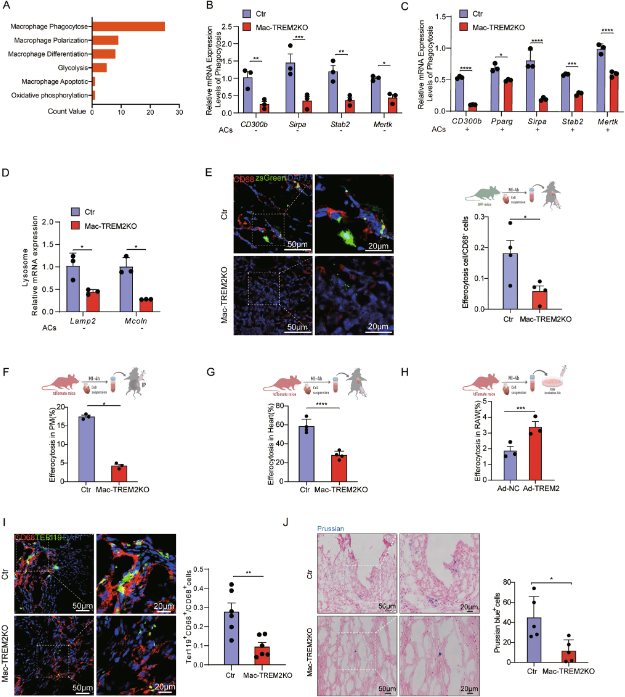
图3:TREM2缺失减弱巨噬细胞的胞葬能力
4. TREM2+巨噬细胞通过胞葬调控SYK-SMAD4信号通路抑制SLC25A53表达
为了深入了解TREM2如何影响巨噬细胞功能的潜在机制,对CCR2cre-EGFP和CCR2cre-EGFP TREM2flox/flox小鼠post-MI第7天的CCR2+细胞进行了RNA-seq。发现在MI发生中,TREM2 KO巨噬细胞经历了几个与吞噬相关过程、碳水化合物代谢相关的转录程序改变(图4A)。此外,SLC25A53被鉴定为TREM2 KO巨噬细胞体外(GSE98563)的差异表达基因,SLC家族与胞葬和体内代谢相关(图4B)。SLC程序在胞葬后被激活,这表明代谢程序发生了转变,在这种转变中改变的代谢物可以影响微环境中的大量细胞。在TREM2 KO BMDM中,ACs刺激下SLC25A53表达增加,而在吞噬抑制剂细胞松弛素D处理后减少,这表明巨噬细胞中TREM2影响SLC25A53的表达(图4C,D)。为了探索TREM2如何调节SLC25A53,用KnockTF筛选潜在的调节因子。结果表明SMAD4是关键的调节因子(图4E),TREM2可能通过激活SYK促进SMAD4的表达。在对照组和TREM2 KO细胞间验证这一假设(图4F)。使用SYK抑制剂或SMAD4抑制剂能够挽救表型,证明SYK-SMAD4-SLC25A53信号通路(图4G)。综上得出结论,TREM2在胞葬后通过SYK-SMAD4抑制SLC25A53的表达。
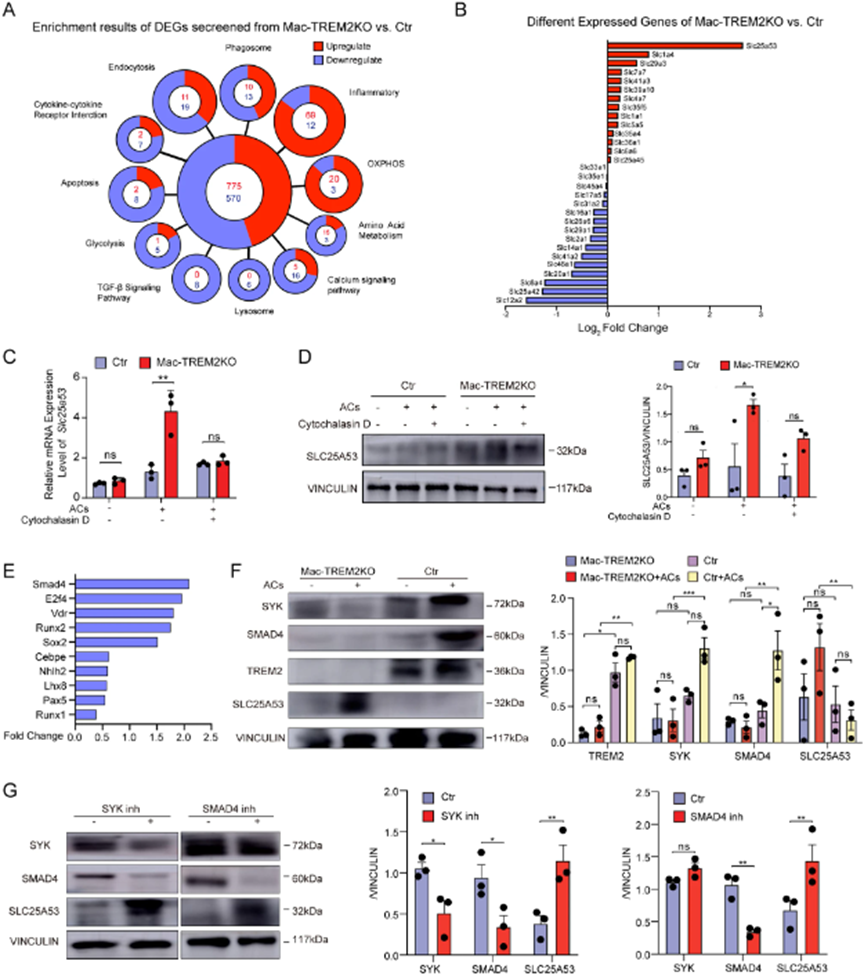
图4:胞葬后TREM2+巨噬细胞通过SYK-SMAD4-SLC25A53途径下调SLC25A53表达
5. TREM2+巨噬细胞通过胞葬后影响线粒体SLC25A53转运NAD+
已有研究结果促使作者进一步研究SLC25A53的功能。该基因有一个能将NAD+导入线粒体哺乳动物转运体中的重要同源物SLC25A51。因此,作者怀疑SLC25A53也可以转运NAD+。为了探索这一假设,首先采用AutoDock Vina模拟SLC25A53和NAD+之间的分子对接,结合能为-8 kcal/mol,表明二者具有很强的真正结合潜力(图5A)。接下来评估了SLC25A53体外转运NAD+的能力。TREM2 KO中,线粒体NAD+水平显著增加,但细胞松弛素D处理后这种增加消失了(图5B)。此外,转染si-SLC25A53后,TREM2 KO线粒体中NAD+水平升高受到抑制(图5C)。在不添加NAD+的情况下,si-SLC25A53组线粒体NAD+水平几乎与NAD+合成抑制剂FK866处理组相当,而外源性NAD+并没有增加si-SLC25A53中线粒体NAD+的含量(图5D)。这表明在TREM2+巨噬细胞中线粒体转运NAD+时SLC25A53起了作用,但对NAD+的整体细胞水平没有影响。
6. TREM2通过SLC25A53促进巨噬细胞中衣康酸生成
研究表明,胞葬后发生代谢重编程,胞葬的代谢副产物可以影响周围的细胞。post-MI Mac-TREM2 KO中ACs减少和纤维化增加,为了调查TREM2+巨噬细胞是否分泌影响缺血微环境内心肌细胞或成纤维细胞的产物。作者先使用坏死心肌细胞的上清液来刺激对照和Mac-TREM2 KO BMDMs,然后收集这些BMDMs的条件上清来刺激缺血和缺氧心肌细胞以及成纤维细胞。值得注意的是,来自TREM2 KO巨噬细胞的上清液处理时观察到心肌细胞凋亡的增加和成纤维细胞增殖的减少(图5E,F)。根据已有的研究结果和文献,提出了一个新假设,即TREM2+巨噬细胞通过胞葬后下调SLC25A53的表达,从而影响糖代谢,其代谢副产物可能对心肌细胞和成纤维细胞产生影响。
为了验证猜想并寻找可能的代谢物,作者将凋亡的H9C2细胞与BMDM一起孵育,然后对分离的线粒体进行LC-MS代谢谱分析。观察到TREM2 KO BMDM中TCA循环代谢物的水平较高(图5G),在分析的代谢物中,衣康酸是最显著的代谢物(图5H)。比较衣康酸衍生物4-OI和衣康酸对缺血和缺氧条件下心肌细胞或成纤维细胞的影响。结果表明4-OI和衣康酸处理组均表现出较高的成纤维细胞增殖(图5I)和较低的细胞凋亡。衣康酸来源于巨噬细胞中TCA循环中的顺乌头酸,这种有机酸被认为是代谢过程中异柠檬酸(ICIT)转化为氧戊二酸(AKG)时潜在断点的结果。此外,干扰NAD+运输进入线粒体后TCA循环中断,这种干扰与导致衣康酸产生的断点相吻合。这表明TREM2+巨噬细胞中的SLC25A53下调通过非活性NAD+运输中断TCA循环导致衣康酸上调(图5J)。
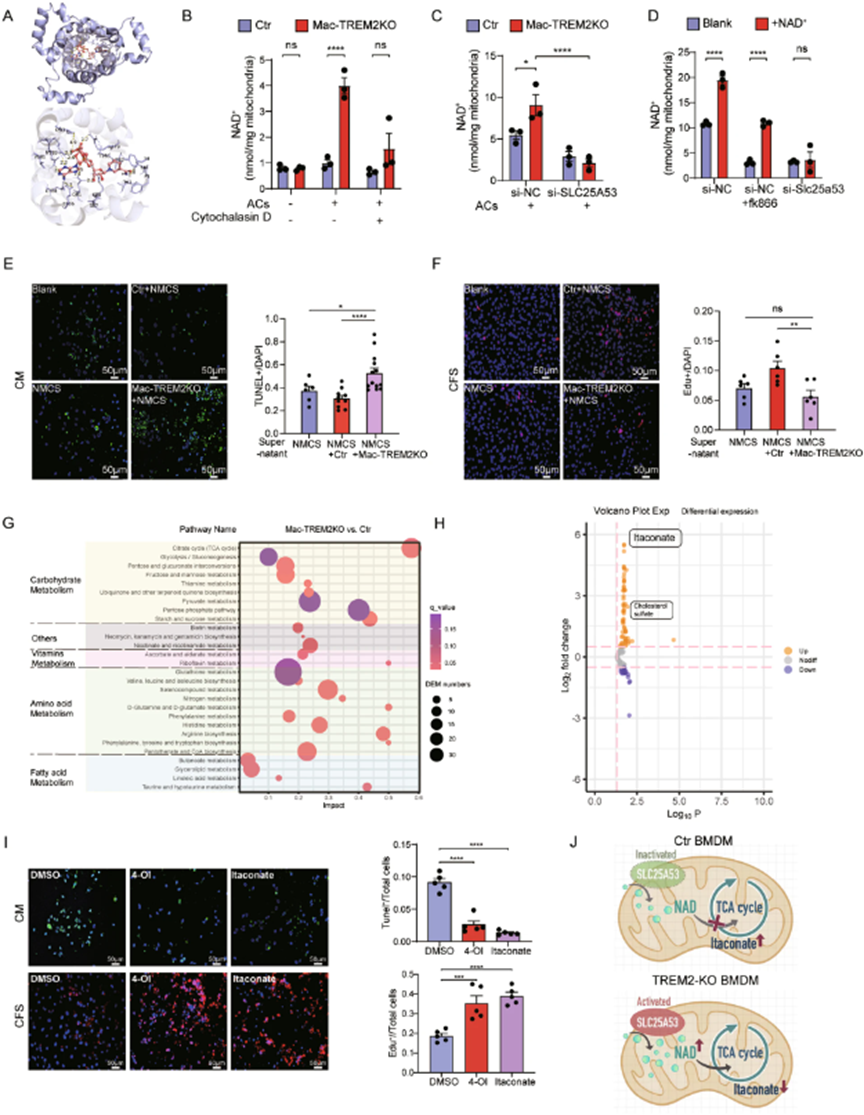
图5:TREM2+巨噬细胞通过SLC25A53影响线粒体NAD+的转运进而促进衣康酸释放
7. 注射TREM2腺病毒后改善左心室重构
小鼠LAD结扎后立即向心肌注射TREM2(Ad-TREM2)腺病毒和对照(Ad-NC)腺病毒来进行挽救实验,以验证TREM2在体内的效果。接受Ad-TREM2治疗的小鼠在post-MI第7天的EF显着提高(图6A)。此外,Ad-TREM2组表现出更好的LV重塑(图6B、C),TREM2表达较高,SLC25A53表达较低(图6D)。此外,研究显示心脏中近80%的Ad-TREM2(GFP+TREM2+)细胞是CD64+巨噬细胞(图6E)。此外,Ad-TREM2小鼠中分离的心脏巨噬细胞中SLC25A53表达水平较低,NAD+含量降低(图6F,G)。这些结果证实了TREM2过表达下调巨噬细胞中的SLC25A53,并改善心肌梗死后心脏的功能和结构。进一步研究了衣康酸对心肌梗死后心脏损伤的作用。结果表明,4-OI注射导致EF显著恢复(图6H)。Masson染色显示4-OI给药的小鼠壁厚增加(图6I,J)。这些结果提示衣康酸衍生物对心肌梗死的潜在治疗作用。.
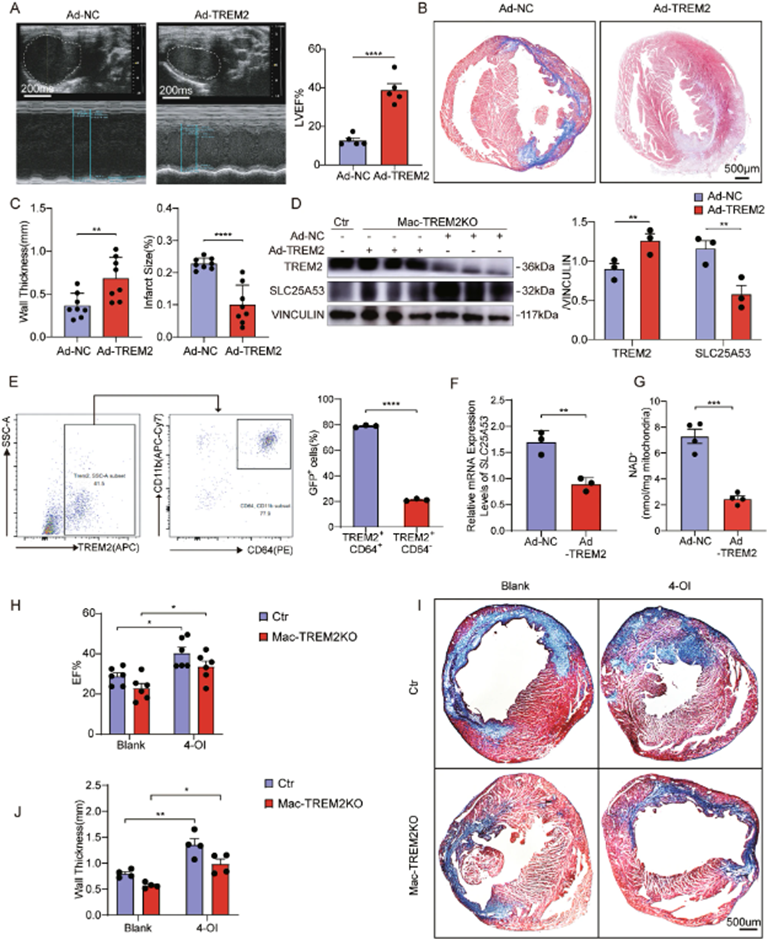
图6:体内注射TREM2-过表达腺病毒后改善左心室重构
结论
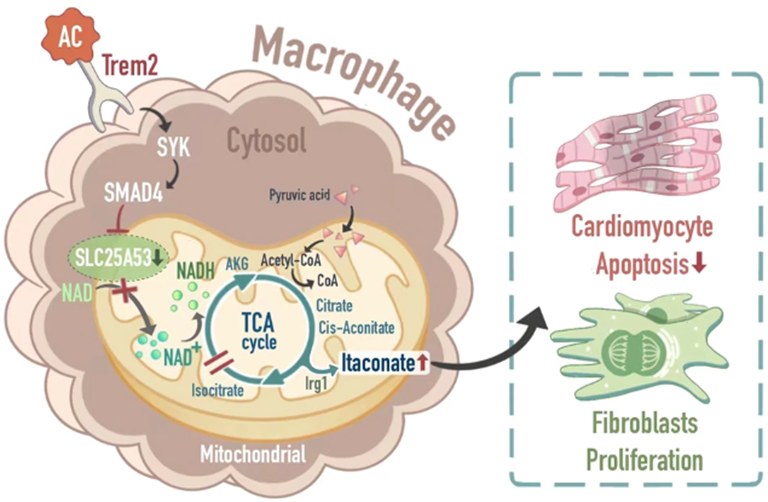
本研究表明心肌梗死中巨噬细胞特异性表达TREM2,使用RNA-seq、蛋白质和分子对接以及靶向代谢组学(LC-MS),发现表达TREM2的巨噬细胞在胞葬后通过SYK-SMAD4信号通路抑制SLC25A53转录,损害NAD+运输到线粒体功能,SLC25A53下调导致三羧酸循环中出现断点,随后增加衣康酸的产生。体外实验证实,TREM2+巨噬细胞分泌的衣康酸抑制心肌细胞凋亡并促进成纤维细胞增殖。相反,在巨噬细胞中过度表达TREM2可以改善心脏功能。机制图如下所示。总之,本研究为TREM2巨噬细胞在心肌梗死中的作用提供了新的见解,并揭示了潜在的机制,为增强损伤后心脏修复开辟了新的可能性。
实验方法
小鼠心肌梗死模型及超声心动图,免疫荧光染色和激光共聚焦荧光显微镜分析,组织学,蛋白提取及蛋白质印迹检测,定量聚合酶链反应PCR,流式细胞术,心脏内注射腺病毒,骨髓来源巨噬细胞的生成,原代心肌细胞和成纤维细胞培养,从hiPSCs分化心肌细胞,原代心肌细胞动作电位的测定,外分泌体外试验,衣康酸刺激,体内和体外的胞葬实验,NAD/NADH+检测,粒体NAD+摄取测定,RNA测序,LC-MS分析
参考文献
Gong Shiyu, Zhai Ming, Shi Jiayun, et al. TREM2 macrophage promotes cardiac repair in myocardial infarction by reprogramming metabolism via SLC25A53.[J] .Cell Death Differ, 2024, undefined: undefined.