






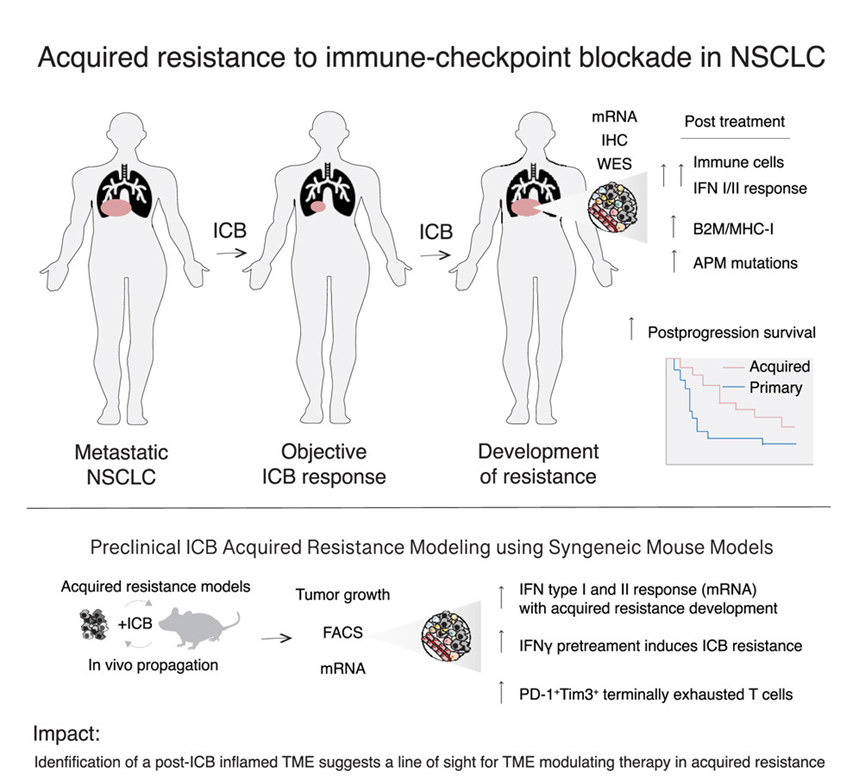
非小细胞肺癌免疫治疗获得性耐药的临床和分子特征
虽然PD-(L)1阻断免疫治疗是肺癌的常规治疗,但对获得性耐药知之甚少。在1201例接受PD-(L)1阻断治疗的非小细胞肺癌(NSCLC)患者中,获得性耐药很常见,发生在>60%的初始应答者中。获得性耐药显示炎症和干扰素(IFN)信号的不同表达。复发肿瘤可以通过IFNγ应答基因的上调或稳定表达来分离。IFNγ应答基因的上调与以持续IFN信号传导、免疫功能障碍和抗原呈递基因突变为特征的推定耐药途径有关,这些特征可以在体外IFNγ治疗后对PD-(L)1阻断获得性耐药的多种小鼠模型中重现。NSCLC对PD-(L)1阻断的获得性耐药与持续但改变的IFN反应有关。获得性耐药的持续炎症而非被排除或遗弃的肿瘤微环境可能为有效重编程和逆转获得性耐药的治疗策略提供信息。本文于2024年1月发表于《Cancer cell》,IF:50.3,Q1。
技术路线:

研究机制
主要研究结果:
1、对PD-1阻断的AR在非小细胞肺癌中很常见
2011年4月至2017年12月期间,在纪念斯隆-凯特琳癌症中心(MSK)接受PD-1阻断治疗的1201例非小细胞肺癌患者中,243例(20%)获得了初步缓解。许多有反应的患者最终发展为AR,使用竞争风险模型随访5年,估计累积AR率为61% (95% CI 36%-85%)(图1A)。AR的发病是可变的(52%在1年内,39%在1 - 2年内,11% > 2年)(图1B)。随着初始反应持续时间的延长,发生AR的相对风险降低(图1C)。
虽然以前没有直接比较AR和原发性耐药,但作者假设这些情况在生物学和临床上是不同的。与此一致的是,作者发现AR和原发性耐药患者的一些基线临床特征存在差异(图1D)。特别是在基线(治疗前)组织中的高肿瘤PD-L1蛋白表达在AR患者中与原发性耐药患者相比更为丰富(55%对28%,Fisher’s p = 0.02)。器官特异性进展模式也有所不同,肝转移是原发性耐药进展的常见部位,但在AR中相对不常见(31%对7%,优势比6.23,Fisher’s p < 0.0001,图1E)。也许最值得注意的是,与原发性进展相比,AR患者的进展后总生存期明显更长(中位18.9个月对4.4个月,log rank p < 0.0001图1F),这可能表明持续的、部分有效的抗肿瘤免疫反应,即使在AR初始发病后也能延长生存期。总的来说,AR在很大程度上以不同的临床特征为特征,这表明AR可能具有不同于原发性耐药的潜在免疫生物学特征,需要专门的分析。
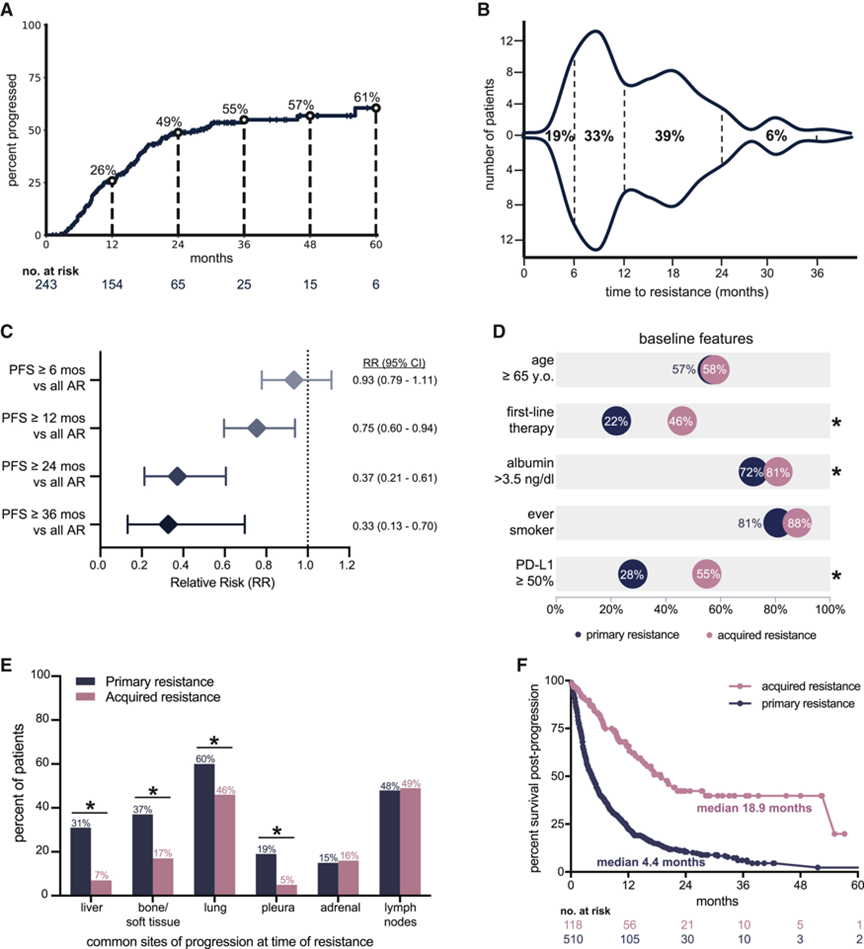
图1 肺癌获得性免疫治疗耐药的临床特点
2、PD-1拮抗剂对AR的分子谱分析的患者队列
为了研究AR对非小细胞肺癌患者PD-1阻断的分子机制,作者从一部分患者治疗前和/或治疗后的肿瘤中获得了基于微阵列的全转录组表达数据和全外显子组测序(WES)数据。经过质量控制和样品优先排序,分子数据的初步分析集中在29例患者的42个肿瘤样本(治疗前13个,治疗后29个)的表达数据和22例患者的34个肿瘤样本(治疗前15个,治疗后19个)的外显子组数据(图2A)。13名患者在治疗前和治疗后的组织中都有表达数据;12名患者在治疗前和治疗后的组织中都有外显子组数据。所有治疗后样本都是在放射学进展到PD-1阻断(从进展到采集样本的中位时间为3.7周,四分位数范围[IQR] 1.8-10.4)和开始新的全身治疗之前获得的(患者未接受PD-1阻断联合化疗;图2B)。
作者和其他人的研究表明,AR经常以低进行性模式发生,这突出了在AR分析中评估病变水平反应的重要性。因此,作者检查了从每个样本中收集的病变水平的反应(和耐药性),以优化治疗前和治疗后样本分别可靠地代表反应性和AR肿瘤的生物学。具体来说,所有治疗后的样本均来自病变特异性放射学反弹生长或新生生长的部位(图2C)。
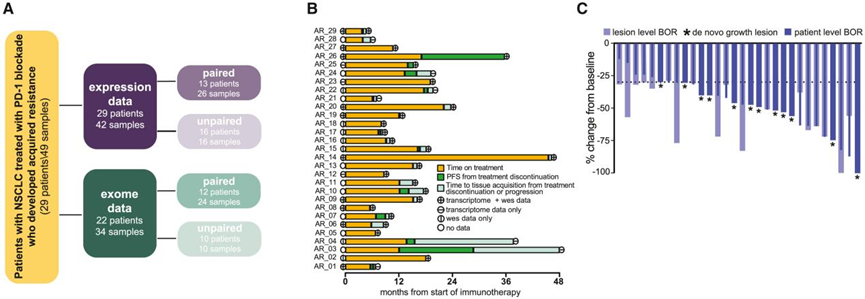
图2 用于外显子组和表达分析的患者队列概述
3、AR对PD-1的阻断与不同的转录谱相关
作者使用单一样本基因集富集分析(ssGSEA)对标志基因集(如之前应用的致癌、细胞应激、免疫、基质和其他过程)进行通路水平评分,总结基因表达值。利用富集评分(ES)对26个配对样本进行的PCA聚类表明,样本根据治疗前和治疗后的配对时间点分离,分离主要由免疫相关标志基因集驱动(图3A和图3B)。标志基因集配对样本的差异表达分析表明,治疗后IFN α / γ应答、氧化磷酸化和DNA修复通路显著上调(错误发现率[FDR] < 0.1,图3C)。对使用CIBERSORT21获得的批量表达的免疫细胞估值进行计算反卷积,基于此对配对样本进行聚类,结果表明治疗前和治疗后样本的分离,尤其是由CD8+ T细胞浸润驱动(图3D和3E)。通过配对的治疗前和治疗后样本的差异分析,治疗后也观察到CD8+ T细胞(FDR < 0.1,图3F)。
一些临床和临床前研究已经生成了大量或单细胞RNA-seq数据集,以鉴定与免疫检查点阻断(ICB)抗性和T细胞功能障碍相关的基因集。其中,与治疗前的样本相比,作者发现AP通路、IFNγ、CD8 T效应细胞和增殖耗竭的CD8+ T细胞的表达增加,而属于WNT通路的基因的表达略有降低(图3G)。与这些与对PD-1阻断的持续免疫应答相关的基因集一致,在治疗后肿瘤中富集的个体基因表达包括GZMA、B2M和CXCL9(图3H)。
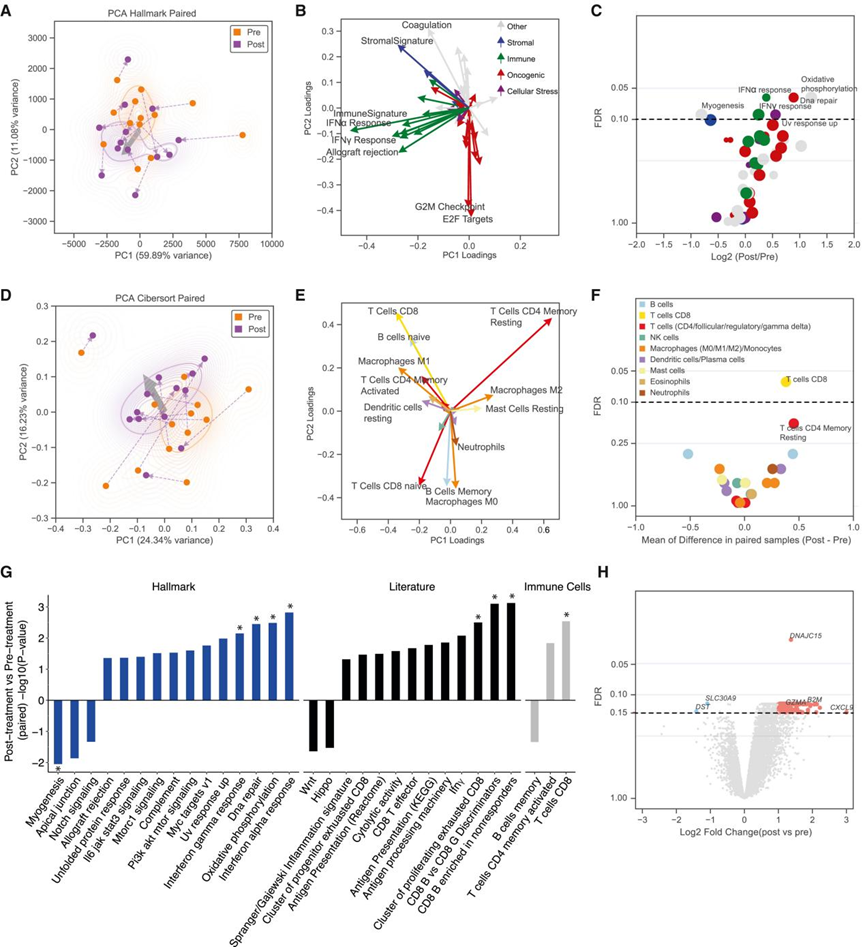
图3 耐药病变表现为IFNγ反应通路上调和CD8+ T细胞浸润
4、IFNγ应答途径的慢性和治疗依赖性增加是AR到ICB的潜在途径
考虑到患者队列中进展时间的可变性(图2B),作者接下来使用Phenopath应用伪时间分析,基于队列中基因表达的可变性,在连续的“潜伏”空间中模拟疾病进展(图4A)。使用治疗前和治疗后样本中基因表达变异性最大的500个基因以及受试者ID和治疗作为协变量,假时间得分通常从治疗前和治疗后样本中增加,特别是对于治疗前假时间估计较低的一组患者(图4B)。为了识别可能与AR相关的通路,作者使用Hallmark和ICB抗性基因集汇编对假时间的变化与通路ssGSEA ES和标签的变化进行了相关性分析(图4C)。在与假时间正相关的前10条通路中,几种IFN I型和II型(IFNγ)特征显著相关,包括构成IFNγ标志基因集的IFN刺激基因(ISGs)(图4D, FDR < 0.01)。值得注意的是,样本可以分为两个子集,大约一半的样本在治疗前后几乎没有增加,另一半的特征是与IFNγ反应相关的ISGs表达升高。因此,作者将患者分为IFNγ反应“稳定”组和IFNγ反应“增加”组(图4D和4E)。
由于在黑色素瘤和其他癌症的临床前小鼠模型中,持续的癌症内在IFN信号传导与ICB耐药有关,作者测试了在作者的临床队列中观察到的ISG特征(IFNα和IFNγ标志基因集)的变化是否与来自ICB耐药黑色素瘤小鼠模型的耐药特征相关。作者发现小鼠来源的ICB耐药特征与治疗诱导的IFNγ反应变化之间存在显著关联(Spearman 's秩相关r = 0.90;p = 2.2e-16;图4F),去除重叠基因后仍然存在(r = 0.86;p = 0.0003)。
与这些患者中IFNγ应答基因的差异变化一致,在数项研究中,IFNγ相关ISGs增加的肿瘤中,与ISGs活化相关的各个转录因子(STAT1和IRF1)以及与CD8+ T细胞耗竭相关的免疫标签(通过标志基因集和文献基因集估计)的推断活性通常增加(图4G-4J)。除了T细胞耗竭的特征外,还观察到调节性T细胞的增加。综上所述,这些数据表明,在非小细胞肺癌中,AR对PD-1阻断的复发模式与肿瘤中IFNγ转录程序的激活、肿瘤特异性IFNγ信号的推定改变(鉴于临床持续的肿瘤生长)以及微环境中CD8+ T细胞耗竭的伴随增加有关。
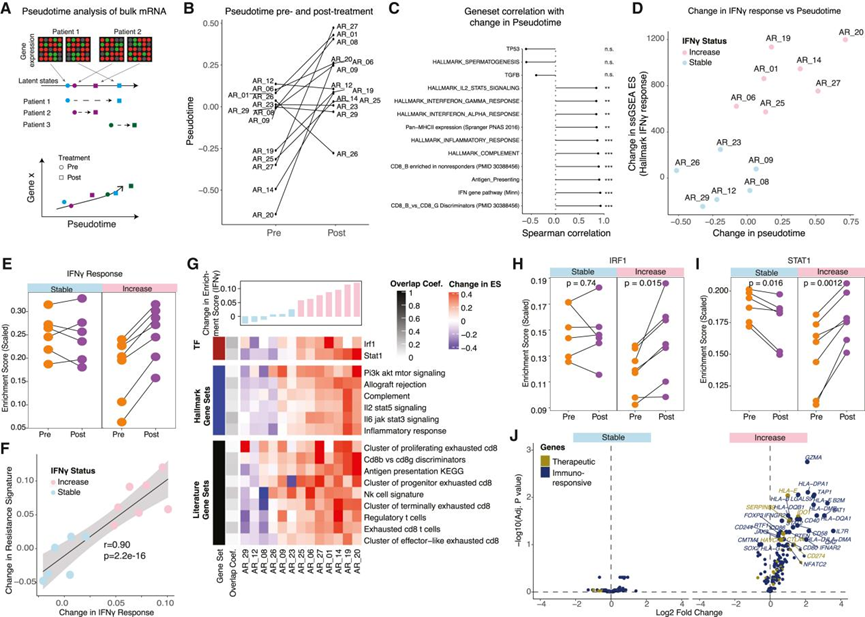
图4 获得性耐药患者的一部分样本在治疗后具有升高的IFNγ反应和T细胞衰竭特征
5、正选择压力对AR中抗原呈递基因突变的影响
非小细胞肺癌的特点是高突变负担,这是免疫治疗反应的一个强有力的预测因素。总体而言,免疫治疗前后的肿瘤突变负荷(Wilcoxon符号秩检验p = 0.6)、已知驱动基因、新抗原负荷、适应度(Wilcoxon符号秩检验p = 0.74)或肿瘤异质性(Wilcoxon符号秩检验p = 0.37)在总体水平上没有显著差异(图5A)。然而,在7例患者样本中有克隆或亚克隆结构重塑的证据。对于其中5个患者样本,克隆突变被保留,而亚克隆突变的子集丢失和/或获得新的亚克隆突变(图5B)。在AR_20治疗后病变中检测到的克隆突变包括STK11基因的无义突变,这与先前观察到的STK11突变与肺腺癌对ICB的耐药性之间的关联一致。一些突变过程,包括外部因素,特别是吸烟,可以影响非小细胞肺癌的体细胞分子谱,并且可以作为突变特征检测。吸烟特征是治疗前病变的主要特征,这些突变在治疗后病变中持续存在。最近的研究表明,在受益于免疫治疗的患者样本中,APOBEC突变特征丰富。在AR_08和AR_20两例患者中,作者观察到与治疗前(AR_08和AR_20分别为5.4%和1.6%)相比,治疗后病变中导致APOBEC突变特征2和13的私有突变比例显著增加(AR_08和AR_20分别为48.3%和14.3%)。
鉴于先前的研究描述了B2M和其他参与AP通路的基因(如TAP1、TAP2和TAPBP)的缺失是耐药肿瘤免疫逃逸的潜在机制,作者进行了一项无偏倚分析,以评估治疗前后个体基因的正选择压力。正如预期的那样,肺癌中的典型驱动突变,如KRAS和TP53,处于强大的正选择压力下,与治疗前相比,治疗后肿瘤中没有明显富集的复发性改变驱动基因(图5C)。然而,在治疗后的AR_14和AR_19肿瘤中分别发现了B2M的无义突变和移码缺失,其他免疫相关基因如IL21R、PDCD5、FKBP1A和FNIP1确实在治疗后富集(图5C)。TAP1、TAP2和TAPBP基因未见潜在致病性突变。
鉴于在ICB耐药肿瘤样本中选择性鉴定出B2M和其他免疫相关基因的突变,作者使用GSEA方法评估了与AP通路相关的其他基因集。具体来说,作者询问是否有证据表明IFNγ选择压力与AP通路失调之间存在关联(图5D)。将具有表达和突变数据的病例的突变变化与IFNγ状态叠加,作者观察到AP通路中的突变富集在IFNγ反应“增加”的患者样本中更为常见,而在IFNγ反应通路“稳定”的患者样本中则相反。值得注意的是,4个克隆或亚克隆结构发生显著变化的患者样本中有3个(AR_20, AR_27, AR_19)在治疗后病变中也显示AP通路基因存在新的突变(图5E)。这4例患者(AR_20、AR_27、AR_19、AR_26)均有可用组织进行肿瘤细胞B2M和1类HLA蛋白表达检测,结果均为阴性或较基线水平下降(图5F和5G)。
为了探索AR在独立队列中的表达模式,作者分析了durvalumab联合tremelimumab在晚期NSCLC患者中进行1b期研究之前获得的原发性和转移性肿瘤样本的RNA-seq数据(“study 06”,NCT02000947)。参加Study 06试验的患者要么是首次接受ICB治疗,要么是先前接受抗pd -(L)1单药治疗失败。没有反应的患者进一步被分类为ICB初级耐药或ICB AR(图5H)。原发性耐药患者从治疗开始≤16周的放射学疾病进展无临床获益证据。AR患者在最初的临床获益(即在任何扫描上完全缓解、部分缓解或疾病稳定)后出现影像学疾病进展。为了研究ICB患者(n = 58)和AR患者(n = 27)之间的表达模式差异,使用ssGSEA使用hallmark和consensusme基因集分析可用的RNA-seq数据,比较肿瘤相关途径的表达模式和从大量肿瘤mRNA估计的免疫细胞浸润。与作者的治疗前后队列相似,研究06中AR患者的样本与未接受治疗的样本相比,骨髓细胞、T细胞和IFNγ相关的ISGs显著富集(FDR < 0.05,图5I)。
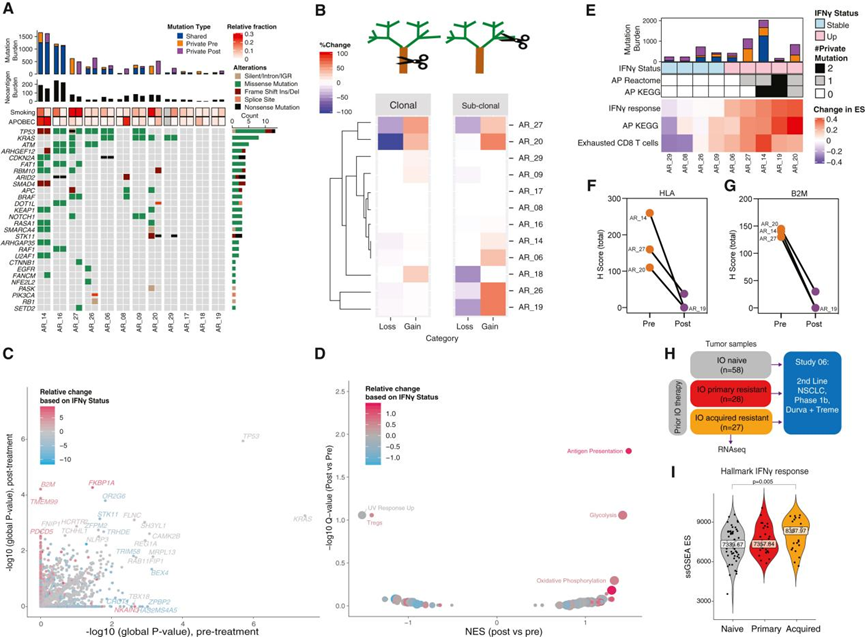
图5 肺癌PD-1阻断获得性耐药的基因组动力学研究
6、AR与ISGs的升高和肿瘤IFNγ信号的改变相关
为了在作者的临床队列中进一步探索与AR到ICB相关的转录特征,作者还使用AR到ICB抑制剂的临床前小鼠模型系统检查了癌细胞的内在转录程序。与对PD-1有反应的人肺癌相似,CT26小鼠模型是致癌物诱导的,具有高肿瘤突变负荷,并且对免疫治疗高度敏感,因此是研究AR的很好的临床前类似物。正如预期的那样,在抗PD-1治疗3周后,皮下CT26肿瘤的肿瘤体积显著减少(图6A)。为了模拟AR,作者切除抗PD-1治疗后的持续活细胞,体外培养,再移植到小鼠体内。这一过程重复了几代,直到CT26肿瘤对抗PD-1抗体治疗不再有反应(图6B)。对来自第二轮(n = 2)和第四轮(n = 4)体内传代肿瘤的ICB耐药癌细胞系进行了大量RNA-seq,并与ICB敏感亲本细胞系(n = 3;图6 c)对比。标记基因集的PCA显示,亲代和第二轮样本倾向于与第四轮样本分开聚类,这种分离主要是由IFN α / γ反应途径驱动的(图6D和6E)。将第四轮样本与亲代样本进行系统比较,发现IFNα和IFNγ反应通路基因显著上调(图6F)。其他生物学过程的基因增加,包括TNFalpha信号(FDR≤0.1)和AP通路(图6I),在第四轮耐药肿瘤细胞中也很明显。与人类数据相似,这些发现表明抗PD -1抵抗与IFNγ相关ISGs的基线表达升高有关。
在第4轮细胞中,随着基线IFNγ反应通路基因的增加,作者还观察到IFN相关转录因子STAT1和IRF1的活性增加(图6G和6H)。这些发现表明,IFN相关转录因子活性的增强可能导致与耐药癌细胞相关的ISG基线表达升高。
为了探究ISGs升高的ICB耐药癌细胞在IFNγ刺激后是否能进一步诱导ISGs,作者用IFNγ刺激细胞系24小时,并与未刺激的对照进行比较。亲代和第二轮细胞系在IFNγ刺激后显示IFN信号相关基因的表达增加,而第四轮细胞系在转录水平上没有显示ISGs的全面诱导(图6J-6L)。此外,IFNγ信号下游的转录因子,如STAT1和IRF1,在第四轮细胞系中表现出相同的模式,IFNγ刺激与对照组之间的表达无统计学差异(图6M和6N)。由于已知IFNγ信号可以上调AP机制通路基因,作者还专门研究了IFNγ对这些基因的影响,进一步支持了观察结果,并显示在第四轮细胞中没有额外诱导IFNγ刺激(图6O)。
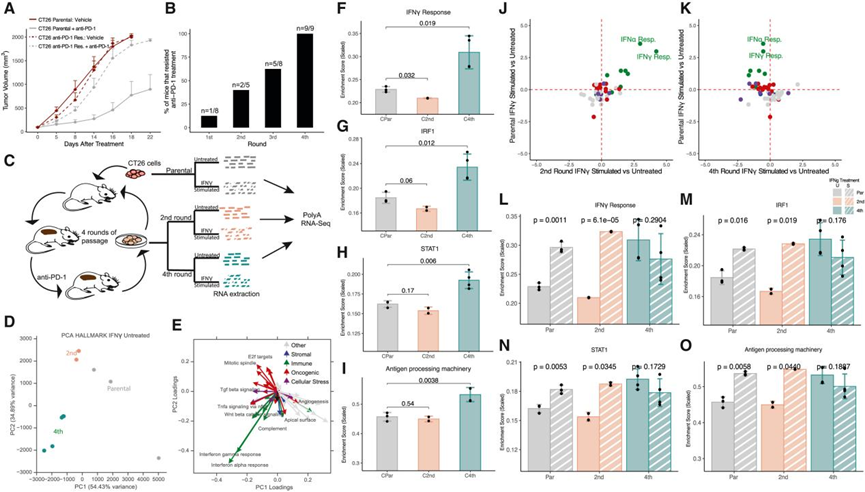
图6 获得性PD-1抗性的小鼠CT26肿瘤细胞系显示IFNγ信号功能失调
7、慢性IFNγ刺激肺癌细胞可促进ISGs升高、耐药和免疫功能紊乱
作者之前已经证明,用IFNγ慢性刺激癌细胞足以使细胞对ICB产生抗性。此外,与抗PD-1 AR的CT26和MC38肿瘤一样,这些慢性刺激细胞增加了一部分ISGs的基线表达和染色质可及性。为了确定慢性IFN刺激是否足以使非小细胞肺癌对ICB产生抗性并促进T细胞功能障碍,作者使用了两种同基因小鼠肺癌模型:Kraslox-stop-lox(lsl)-G12D/+;Trp53flox/flox (KP)基因工程小鼠模型和Lewis肺癌(LLC1)(图7A)。在这两种模型中,将ICB后自发复发的肿瘤与植入小鼠前在体外用低水平IFNγ处理3-4周的癌细胞衍生的肿瘤进行比较(图7B)。通过体外慢性IFNγ刺激3-4周(γLLC1)或晚期复发衍生肿瘤细胞系(ResResLLC1),与慢性IFN信号相关的肿瘤对双ICB(抗PD -1 +抗CTLA -4的组合)的反应减弱(图7B)。这些临床前数据证实了作者之前的发现,即高ISGs与小鼠肿瘤模型中ICB后的进展有关。在含有慢性IFN信号(γLLC1)或晚期复发源LLC1肿瘤细胞系(ResResLLC1)的LLC1模型中,ICB疗效降低的特征是ICB后肿瘤中功能失调的PD-1+TIM-3+耗尽T细胞的积累(图7C和7D)。然后,作者系统地表征了LLC1肿瘤中的免疫浸润(图7E和7F)。采用无监督聚类方法将肿瘤浸润免疫细胞分为12个元簇。有趣的是,主导T细胞衰竭标志物(PD-1+, TIM-3+等)的元簇5和6在慢性IFN刺激或晚期复发的肿瘤样本中显示ICB后频率增加,但在最初敏感的肿瘤样本中没有(图7G, 7H)。
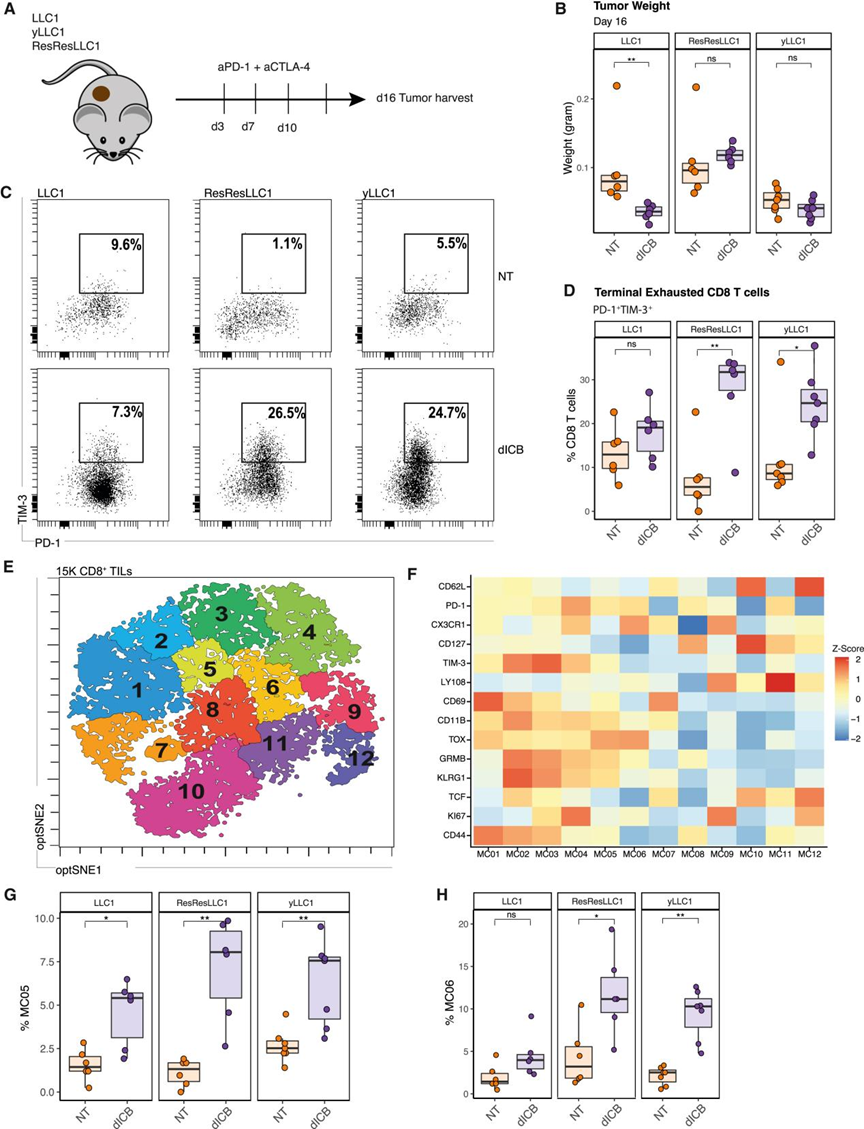
图7 在LLC1同基因肺癌小鼠模型中,免疫检查点阻断获得性抵抗(ICB)与诱导终末耗尽CD8+ T细胞相关
结论:
通过本文提出的多种小鼠获得性耐药模型,作者总结了获得性耐药是如何与癌症内在IFNγ反应上调以及最终肿瘤对有效抗肿瘤免疫的不敏感相关的。另外,作者还表明,体外预处理暴露于IFNγ会导致体内对ICB治疗产生耐药性。此外,作者初步观察到,体内产生的获得性耐药细胞系通常会改变ISG反应,因为与受IFNγ刺激的亲本细胞相比,体外IFNγ刺激与相对较低的ISG激活相关。需要进一步的工作来确定免疫细胞和肿瘤细胞中响应IFNγ信号动力学的具体机制缺陷。总的来说,这些数据可以进一步指导更合理的指导治疗策略,预防、克服和逆转肺癌患者对PD-1阻断的AR。
实验方法:
从MSK队列中生成分子数据集;来自MSK队列的基因表达谱;从表达数据估计基因集富集分数;拟时间分析;MSK队列的全外显子组测序;肿瘤异质性和克隆性;肿瘤外显子组数据中体细胞特征的估计;突变数据中的选择压力分析;系统发育树重建;新抗原预测和适应度评分;研究06样本队列和转录组学分析;抗PD-1耐药CT26肿瘤的产生;抗PD-1耐药CT26细胞系的转录组学和ATAC-Seq分析;抗PD-1耐药MC38肿瘤的产生;抗PD-1耐药MC38细胞系基因表达谱分析;LLC1和KP肿瘤细胞系的生成;分选小鼠肿瘤细胞的RNA-seq生成和分析;LLC1小鼠模型的体内小鼠淋巴细胞研究;流式细胞术特征聚类。
参考文献:
Memon D, Schoenfeld AJ, Ye D, Fromm G, Rizvi H, Zhang X, Keddar MR, Mathew D, Yoo KJ, Qiu J, Lihm J, Miriyala J, Sauter JL, Luo J, Chow A, Bhanot UK, McCarthy C, Vanderbilt CM, Liu C, Abu-Akeel M, Plodkowski AJ, McGranahan N, Łuksza M, Greenbaum BD, Merghoub T, Achour I, Barrett JC, Stewart R, Beltrao P, Schreiber TH, Minn AJ, Miller ML, Hellmann MD. Clinical and molecular features of acquired resistance to immunotherapy in non-small cell lung cancer. Cancer Cell. 2024 Jan 9:S1535-6108(23)00441-5. doi: 10.1016/j.ccell.2023.12.013. Epub ahead of print. PMID: 38215748.