






NRF2通过与TOPBP1协同激活ATR-CHK1信号通路促进辐射抵抗
肺癌是一种致命的恶性肿瘤,每天死亡人数超过350人。临床研究表明约77%的肺癌患者接受放疗(Radiotherapy,RT)。尽管放疗的疗效显著改善,但癌细胞在连续暴露于电离辐射(Ionizing Radiation,IR)时产生抗性导致疾病复发。IR主要通过引起DNA损伤,特别是双链断裂(DSBs)来杀死癌细胞。辐射抗性癌细胞已经发展出强大的DNA损伤修复能力来生存IR。核因子‑红细胞2相关因子2(Nuclear Factor Erythroid 2-related Factor 2,NRF2)与辐射抗性相关。在本研究中,作者采用TCGA数据库和组织芯片分析NRF2与肺癌患者预后的相关性,构建耐辐射肺癌细胞,通过体内和体外实验探讨NRF2在抗辐射中的作用,并采用免疫沉淀、免疫荧光和染色质级分提取等方法探讨其潜在机制。本研究证明NRF2通过与RPA32和TOPBP1合作激活ATR-CHK1信号通路增强辐射抗性,还验证了NRF2可作为放疗的潜在靶点。
该研究于2024年1月1日发表在《Theranostics》,IF:12.4
技术路线
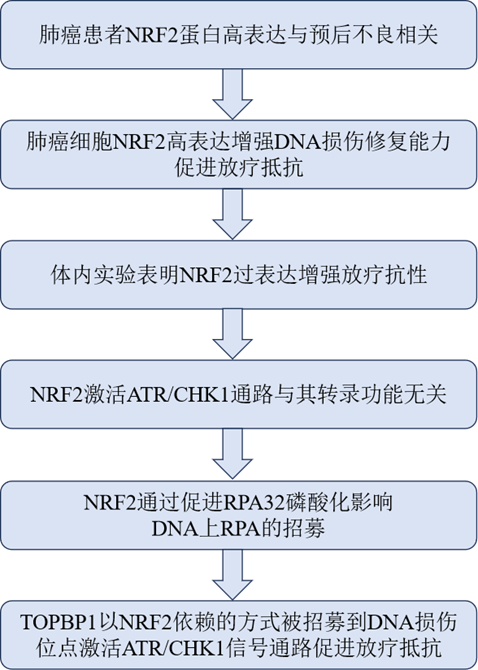
主要研究结果
1.NRF2/KEAP1基因改变引起NRF2激活与肺癌患者预后不良相关
作者通过c-BioPortal网站分析了肺癌患者NRF2/KEAP1基因的变异类型。在分析了来自29项研究的12,478个样本/ 9,930名肺癌患者后,发现NRF2和KEAP1基因的总变异分别为4%和13%(图1A)。遗传突变是NRF2和KEAP1基因变异的主要形式。在拷贝数变异类型中,扩增是NRF2的主要改变类型,而KEAP1的主要改变类型是缺失(图1A)。肺癌的不同亚型,如肺腺癌(LUAD)和肺鳞状细胞癌(LUSC),具有不同的细胞变异类型,导致不同的生长特征。此外,作者发现NRF2的扩增和突变主要发生在LUSC(图1B),KEAP1基因变异在LUAD中发生更频繁(图1C)。接下来通过RNA-seq分析表明,LUAD和LUSC中NRF2的主要CNV类型是扩增和增益,这导致NRF2 mRNA表达增加(图1D-E)。KEAP1的主要变异类型是浅缺失,伴随着较低的KEAP1 mRNA表达(图1F-G)。此外还调查了NRF2/KEAP1基因改变组和NRF2/KEAP1基因未改变组患者之间的临床结果。结果表明,NRF2/KEAP1基因的基因改变与较短的总生存期相关(图1H-I)。NRF2/KEAP1基因改变患者的总生存期中位数分别为38.24和38.14个月,而无基因改变组分别为56.35和54.34个月。因此,上述结果表明,NRF2/KEAP1基因变异导致NRF2表达水平升高,并与肺癌患者预后不良有关。
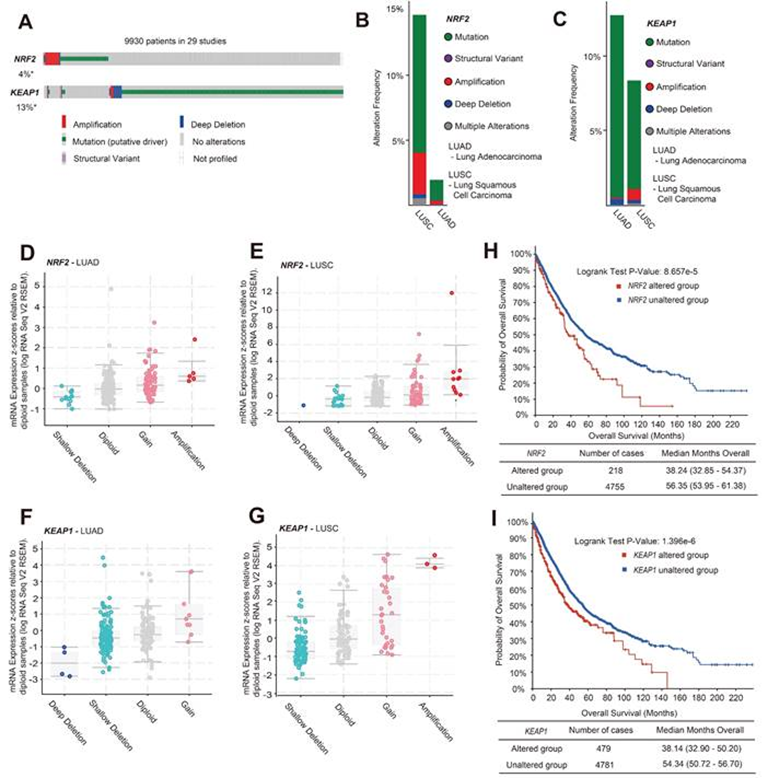
图1:NRF2/KEAP1变异导致的NRF2过表达与肺癌患者预后不良相关
IHC检测由LUAD患者和邻近非癌组织作为对照的肺癌组织样本组成的肿瘤微阵列(TMA)中NRF2蛋白的表达。用NRF2抗体染色TMA,并根据染色强度评估评分。根据细胞质中NRF2染色强度将LUAD样本分为高/中/低NRF2表达水平的三组(图2A)。在LUAD组中,44%的患者属于NRF2高表达组(图2B),该组患者的中位寿命为31.23个月(图2C)。接下来作者根据NRF2蛋白在细胞核中的定位将患者分为阳性和阴性组,结果显示核NRF2检测阳性的患者的寿命明显短于检测阴性的患者(图2D-E)。
总体而言,TMA结果与TCGA数据库分析结果一致,表明肺癌患者NRF2蛋白高表达与预后不良相关。
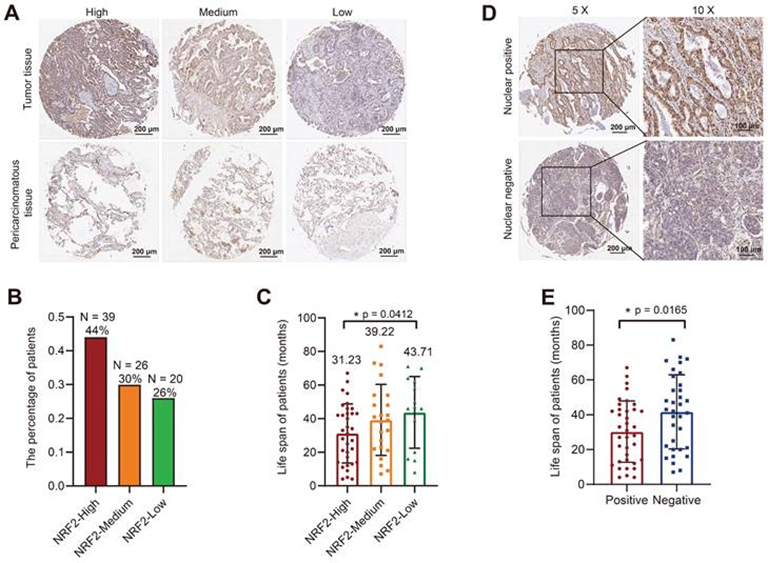
图2:组织芯片染色NRF2与LUAD患者预后的相关性分析。
2.放疗抵抗细胞中NRF2高表达
作者建立了人类肺癌抗辐射细胞并通过集落形成实验进行验证。对肺癌细胞和肺癌放疗抵抗细胞使用质谱进行蛋白质组学分析,发现氧化应激信号通路发生变化。检测了辐射抵抗细胞中NRF2的蛋白水平,发现与A549细胞相比,A549R细胞中具有抗氧化应激功能的NRF2蛋白及其下游血红素氧合酶-1(HO-1)显著上调(图3A)。NRF2向细胞核转移是激活下游基因的先决条件,这些基因编码一系列II期解毒或抗氧化酶。核质分离结果显示,与H460和H1299细胞相比,H460R和H1299R细胞核中NRF2蛋白水平显著升高(图3B-C)。此外,免疫荧光实验显示,在H1299R细胞中NRF2荧光信号更强烈(图3E)。这些结果表明NRF2在辐射抗性细胞中上调,并优先积累在细胞核中。
有研究报道过NRF2可通过激活ATR/CHK1通路导致DSBs和G2/M细胞周期停滞。因此作者接下来监测了放射抗性细胞中ATR信号级联的变化。WB结果显示,IR诱导H460和H1299细胞中ATR和CHK1的磷酸化,H460R和H1299R细胞中ATR和CHK1的磷酸化水平明显更高(图3F-G)。采用免疫荧光监测γ-H2AX以评估DNA损伤。红外线照射后,A549R细胞γ-H2AX病灶的数量显著低于A549细胞,表明辐射抵抗细胞的DNA修复能力优于对照细胞(图3H)。H460和H1299细胞也观察到类似的趋势(图3I-J)。上述结果表明,辐射抵抗细胞中高水平的NRF2蛋白增强了DNA损伤修复能力,这可能是肺癌细胞可以发生辐射抵抗的原因之一。.
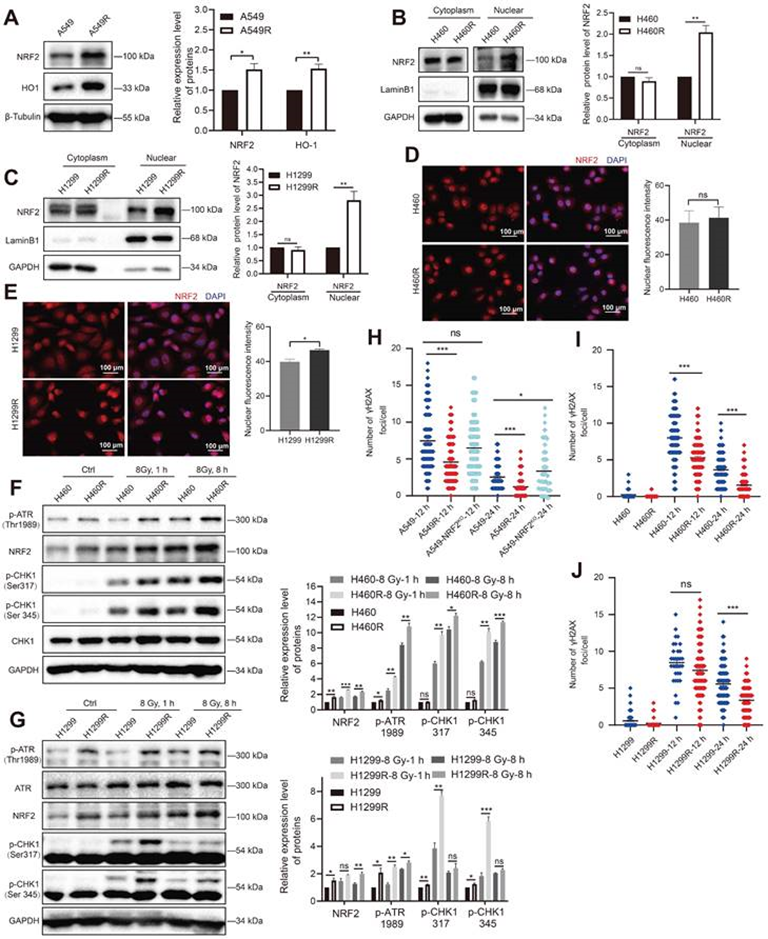
图3:NRF2在抗辐射细胞中表达显著
3. 体内NRF2过表达的抗辐射细胞对放疗的抗性更强
将稳定表达GFP的A549和A549R细胞注射到裸鼠尾静脉,6周后成功构建肺肿瘤转移模型,将小鼠分为对照组(A549)、抗辐射细胞组(A549R)、对照照射组(A549-IR)、抗辐射细胞照射组(A549R-IR)四组。每周用2或4 Gy分次剂量的γ射线局部照射肺部,治疗持续3周(图4A)。荧光信号检测到肺部肿瘤转移(图4B)。与A549组相比,A549-IR组放疗后肺肿瘤转移数量明显减少。相比之下,在注射A549R细胞的小鼠中,接受或不接受放疗的肺转移数量没有观察到统计学上的显著差异,提示A549R肿瘤对IR更具抗性(图4C-D)。结果显示A549R细胞在体内也具有抗辐射性。肺组织切片IHC分析显示,A549细胞源性肿瘤中的Ki67阳性染色在照射后显著下降,但A549R-IR组与A549R-未照射组的Ki67丰度差异较小,提示IR可能不能有效抑制A549R细胞的增殖(图4E)。此外,用IHC检测NRF2的染色强度。A549组的NRF2染色在照射后显著下降,而A549R组的变化不显著(图4F)。总的来说,这些研究结果表明NRF2蛋白与体内肺癌细胞的辐射抵抗之间存在关联。
接下来,作者通过Nrf2+/ -C57BL/6小鼠杂交得到Nrf2+/+和Nrf2-/-幼鼠,以进一步探索NRF2在体内响应IR中的作用(图4G)。将Nrf2+/+和Nrf2-/-小鼠暴露于两种剂量的IR(7.2和6.5 Gy)(图4H),发现Nrf2-/-小鼠表现出更高更快的死亡率(图4I和4K)。Nrf2-/-小鼠的平均体重在全身照射(TBI)后继续下降;而,Nrf2+/+小鼠的平均体重在TBI后大约20天趋于稳定(图4J和4L)。这些结果表明,缺失NRF2基因导致辐射暴露后小鼠死亡率较高,这意味着NRF2可能在体内对辐射反应中发挥保护作用。
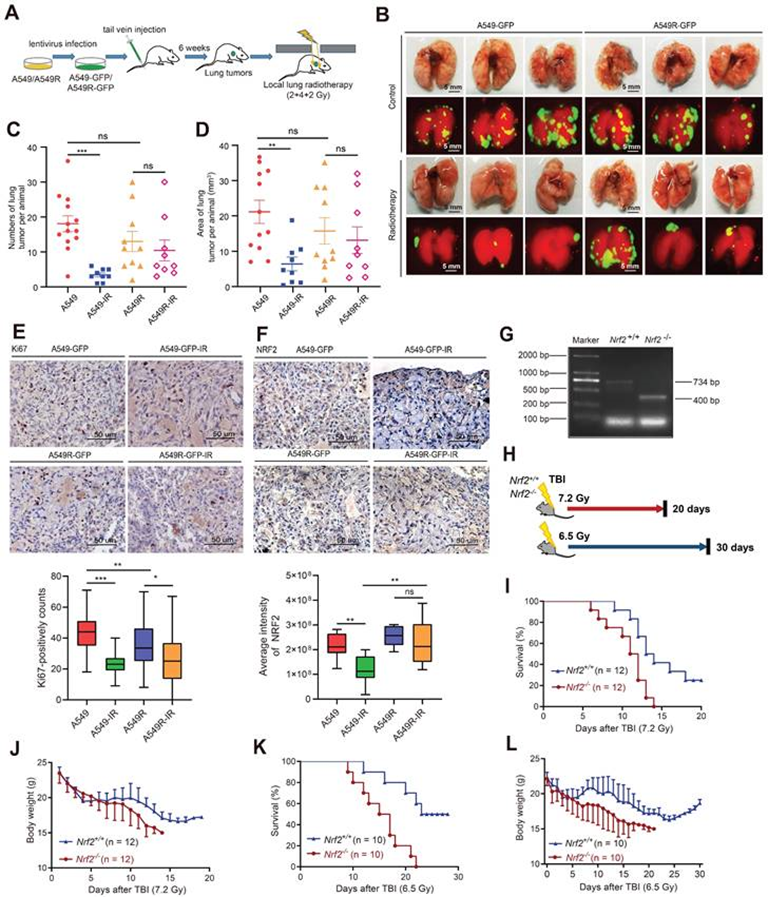
图4:NRF2过表达的放射抗性细胞在体内对放疗的抗性更强
4. NRF2激活ATR/CHK1通路独立于其转录功能
接下来,作者试图阐明NRF2导致放射抗药性的机制。在A549和H1299细胞中,照射以剂量依赖性的方式增加NRF2蛋白水平(图5A-B);8 Gy照射以时间依赖性的方式增加NRF2蛋白水平(图5C-D)。之前的实验结果表明NRF2蛋白在肺癌组织细胞核中的积累与肺癌预后较差有关(图2D-E),抗辐射细胞细胞核中的NRF2蛋白水平也较高(图3B-C)。因此,从细胞中分离出染色质,并检查照射后NRF2与染色质的结合。结果表明,照射后NRF2蛋白与染色质的结合显著增加(图5E),这表明NRF2在红外下转移到细胞核并与染色质结合。此外,通过免疫荧光研究NRF2的亚细胞定位。结果表明,NRF2被招募到激光微照射引起的DNA损伤位点,并在H1703和A549细胞中与γ-H2AX共定位(图5F)。
NRF2作为一种转录因子在应激条件下调节BRCA1和53BP1的表达。BRCA1和53BP1都是DNA损伤反应过程中的关键蛋白。在A549细胞中敲低NRF2后53BP1和BRCA1蛋白的水平显著下降;而53BP1和BRCA1的敲低并不影响CPT处理后ATR和CHK1的激活(图5G)。这些结果表明53BP1和BRCA1可能不是NRF2/ATR/CHK1信号通路激活的介导者。为了确认NRF2促进ATR-CHK1信号通路的激活不依赖于其转录调节功能,作者构建了删除Neh1结构域的NRF2突变质粒(Δ434-561-NRF2)(图5H),并在A549-NRF2 KO细胞中分别表达HA-NRF2和Δ434-561-NRF2。当细胞用8 Gy IR处理时,HA-NRF2和Δ434-561-NRF2的表达都显著促进ATR和CHK1磷酸化(图5I)。这些结果表明NRF2可以保持独立于转录功能的基因组稳定性。
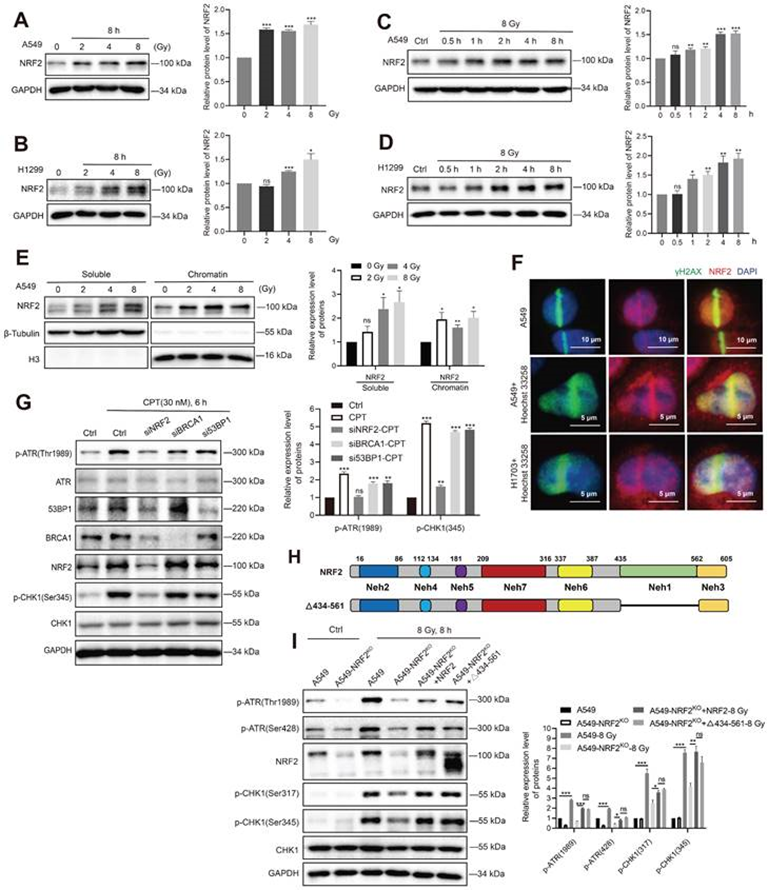
图5:NRF2激活ATR/CHK1通路独立于其转录功能
5. NRF2促进RPA32磷酸化和RPA在ssDNA的积累
NRF2可以直接结合并激活ATR,而RPA调节ATR激活。因此,作者探讨了NRF2是否可以影响RPA在DNA损伤过程中的作用。RPA70和RPA32是RPA复合物的两个主要亚基。在照射后6、12和24小时检查了用siNRF2处理或未处理的A549细胞中RPA32和RPA70病灶的数量。免疫荧光结果显示,敲低NRF2后A549细胞中RPA70病灶阳性细胞的比例明显高于A549细胞暴露于IR后(图6A)。同样,敲低NRF2降低了细胞暴露于IR后RPA32病灶阳性细胞的比例(图6B-C)。DNA结合的RPA与许多蛋白质相互作用以调节DNA代谢,RPA封装的ssDNA也是复制应激诱导的DNA损伤反应过程的平台。接下来,提取染色质结合蛋白以确定NRF2是否影响RPA32蛋白与DNA的结合,发现NRF2的敲除在照射后显着抑制RPA32蛋白与染色质的结合,但不影响RPA32的可溶性形式(图6D)。结果暗示NRF2可能有助于RPA32与染色质的结合在DNA损伤条件下形成病灶。
进一步研究了NRF2是否会影响RPA32的磷酸化。在A549细胞中,NRF2的敲低降低了CPT处理后RPA在Ser33和Thr21的磷酸化(图6E)。此外还观察到RPA32(Ser33、Thr21和S4/S8)的磷酸化水平在照射后10小时内持续增加,且A549R细胞中RPA32的磷酸化增加更加明显(图6F)。NRF2敲除细胞中RPA32的磷酸化明显低于野生型细胞,并且通过在CPT(图6G)处理后将Flag-NRF2重新引入A549-NRF2KO细胞来挽救这一缺陷。因此,研究结果表明NRF2通过促进DNA损伤过程中RPA32的磷酸化来影响DNA上RPA的招募。
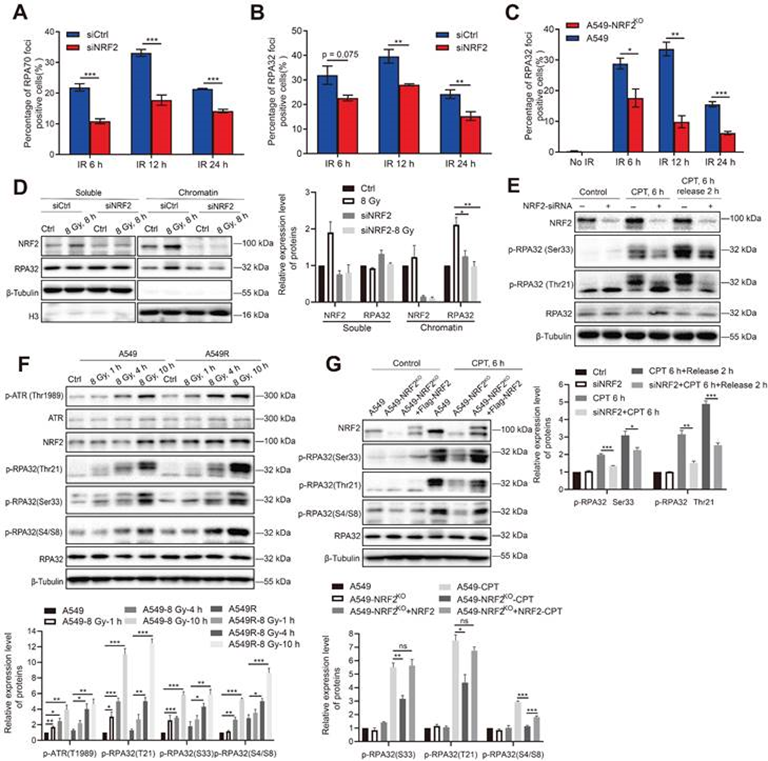
图6:NRF2促进RPA32的磷酸化和RPA在ssDNA的积累。
6. NRF2协同TOPBP1激活ATR/CHK1信号通路
RPA32和CHK1都是ATR的磷酸化底物。有研究表明,TOPBP1和ETAA1的激活对应于不同的ATR功能。ETAA1在正常和应激复制过程中负责ATR介导的RPA磷酸化,而TOPBP1激活ATR/CHK1信号通路以响应复制应激。NRF2、TOPBP1和ETAA1在A549细胞中分别敲除,以比较NRF2、TOPBP1和ETAA1之间的差异。结果表明,转染siNRF2的A549细胞在8Gy照射后ATR和CHK1的磷酸化水平比转染siTOPBP1和siETAA1的细胞更显著降低,表明NRF2在ATR的磷酸化中发挥了重要作用(图7A)。而与单独敲除NRF2相比,同时敲除TOPBP1和NRF2并没有进一步降低ATR磷酸化水平(图7A)。这些观察结果表明NRF2可能与TOPBP1在同一途径中发挥作用。因此,构建的Flag-TOPBP1和HA-NRF2融合蛋白进行验证,用60 nM CPT处理细胞后,siTOPBP1在H1299R细胞中ATR/CHK1没有磷酸化(图7B)。表达HA-NRF2没有挽救这一缺陷,这表明TOPBP1在NRF2/ATR/CHK1途径中的关键作用(图7B)。同时,Flag-TOPBP1的表达也未能磷酸化ATR和CHK1。
TOPBP1通过与RAD9-HUS1-RAD1(9-1-1)的复合物被招募到DNA损伤位点,并通过其AAD样区域结合介导ATR的磷酸化。在染色质结合蛋白检测实验中,WB显示NRF2的下调显著抑制了TOPBP1蛋白与辐照A549细胞染色质的结合(图7D)。使用靶向HPRT基因的Cas9/sgRNA实现了位置特异性DNA双链断裂,以进一步证实NRF2在TOPBP1招募到DNA损伤位点中的作用。荧光显微镜显示TOPBP1与A549细胞中的γH2AX共定位。NRF2缺乏显著减弱了A549-NRF2KO细胞中TOPBP1病灶的强度(图7E-F)。图7G结果清楚地表明,A549细胞中激光微辐射后TOPBP1被招募到DNA损伤位点,但在A549-NRF2KO细胞中没有。这些结果表明,NRF2可以影响DNA损伤期间TOPBP1招募。接下来使用Co-IP验证DNA损伤中TOPBP1和NRF2之间的相互作用。从A549细胞的染色质和可溶性提取物中免疫沉淀NRF2后可检测到TOPBP1。此外,在IR后细胞的染色体提取物中TOPBP1和NRF2之间的相互作用增强(图7H)。这些结果表明NRF2与TOPBP1合作促进ATR/CHK1信号通路的激活,并对照射后TOPBP1向DNA损伤位点的募集有促进作用。
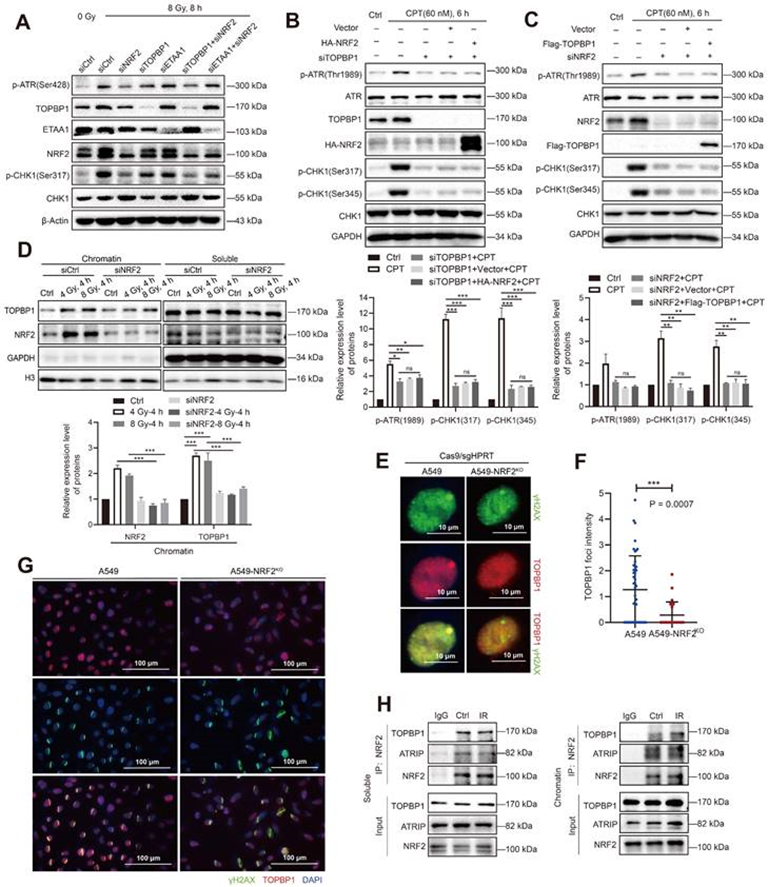
图7:NRF2协同TOPBP1激活ATR/CHK1信号通路
结论
作者证明NRF2的高核表达与肺癌患者的不良预后相关,并有助于辐射抗性的发展。响应IR或CPT,NRF2被转移到DNA损伤位点,促进RPA32的磷酸化,并通过招募TOPBP1到DNA损伤位点激活ATR/CHK1通路(图8)。这项研究表明NRF2可能是改善肺癌放疗的一个有希望的靶点。
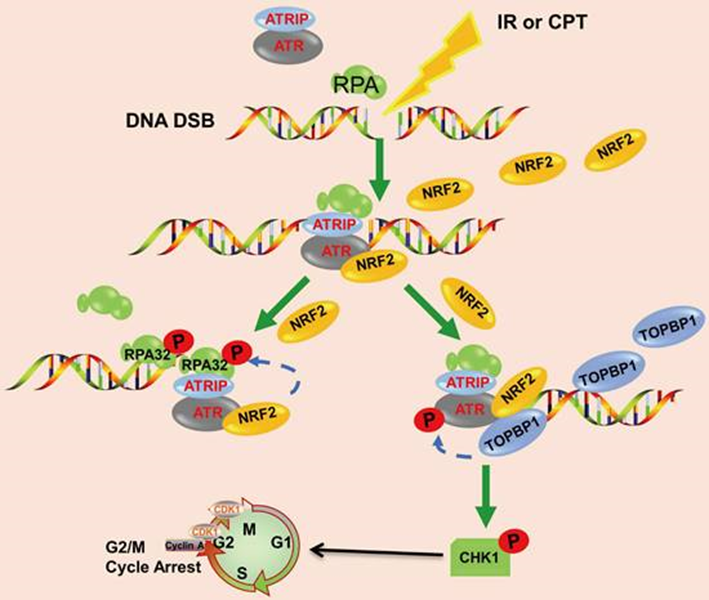
图8
实验方法
细胞培养和药物治疗,组织微阵列,辐射抗性细胞系构建,全细胞裂解物收集和蛋白质印迹法,免疫荧光分析,Co-IP实验,活性氧水平的测定,集落形成试验,肿瘤球形成实验,核和细胞质蛋白质提取,提取染色质部分,慢病毒转染实验,siRNA转染实验,质粒转染实验,小鼠实验,免疫组织化学染色,TUNEL实验
参考文献
Sun Xiaohui, Dong Mingxin, Li Jiale, et al. NRF2 promotes radiation resistance by cooperating with TOPBP1 to activate the ATR-CHK1 signaling pathway. [J]. Theranostics, 2024, 14: 681-698.