






circKIF4A-miR-637-STAT3的新轴促进三阴性乳腺癌的脑转移
在三阴性乳腺癌(TNBC)患者中,远处转移是导致死亡的主要原因。我们之前的研究表明,circKIF4A极大地促进了TNBC的进展,但其在TNBC脑转移中的作用和分子机制仍然不确定。在这项研究中,我们发现circKIF4A在TNBC细胞系和脑转移中显著上调。抑制circKIF4A会损害TNBC的增殖、迁移和引起脑转移的能力。荧光素酶报告基因检测证实circKIF4A与STAT3 3’UTR竞争miR-637的结合。Western blot分析显示,抑制circKIF4A可降低STAT3和p62的表达,同时增加LC3B-II/ LC3B-I比值和Beclin的表达,表明circKIF4A下调通过与STAT3竞争miR- 637结合而诱导自噬。通过采用竞争性内源性RNA (ceRNA)机制,circKIF4A-miR-637-STAT3轴协调TNBC中的脑转移。因此,circKIF4A可以作为TNBC脑转移的预后生物标志物和治疗靶点。本文于2023年11月发表于“Cancer Letter”(IF=9.7)上。
技术路线:
结果:
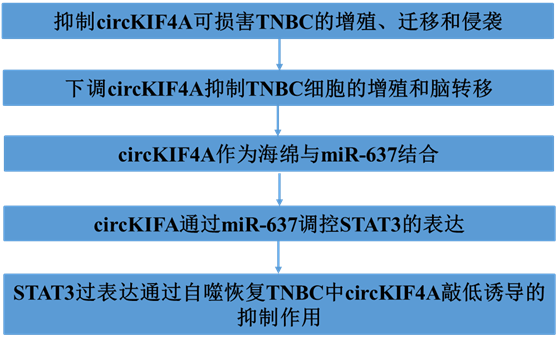
1)抑制circKIF4A可损害TNBC的增殖、迁移和侵袭
为了探索KIF4A衍生的环状RNA hsa_circ_0007255 (circKIF4A)在TNBC脑转移中的功能,我们采用qRT-PCR验证了circKIF4A在脑转移和原发性乳腺癌组织中的表达。22例脑转移患者显示circKIF4A水平升高,且在脑转移组织中明显升高(图1A)。然后我们在TNBC细胞系和MCF10A中进行RT-qPCR检测,发现circKIF4A在TNBC细胞系中上调(图1B)。用RNase R(核糖核酸酶R)处理HCC1806和SUM159PT细胞株RNA样品后,我们使用qRT-PCR检测了两株细胞株中circKIF4A及其亲本基因编码KIF4A mRNA和GAPDH的表达水平。结果表明,与线性KIF4A mRNA和GAPDH相比,circKIF4A对RNase R处理具有更强的耐受性,这与环状RNA的特征一致(图1C)。我们设计了两个siRNA来探索circKIF4A的功能,并通过RT-qPCR在TNBC细胞中验证了其敲低作用(图1D)。采用CCK-8和集落形成法检测TNBC细胞的活力,结果表明,抑制circKIF4A可显著损害TNBC细胞的增殖能力(图1E, F, I)。此外,在伤口愈合和transwell试验中,敲低circKIF4A可抑制TNBC细胞的迁移和侵袭能力(图1G-I)。这些结果表明,抑制circKIF4A会损害TNBC细胞的增殖、迁移和侵袭能力。
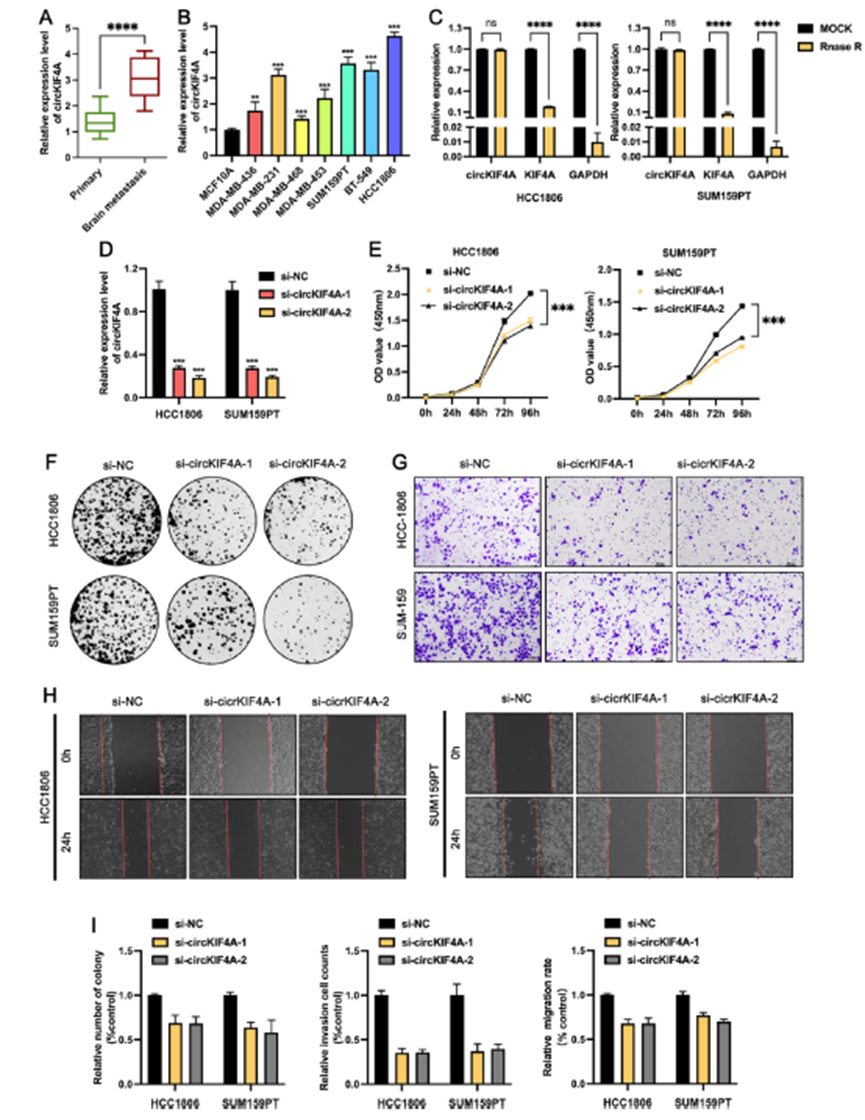
2)下调circKIF4A抑制TNBC细胞的增殖和脑转移
在通过体外细胞实验证实了circKIF4A在TNBC细胞中的促瘤作用后,我们利用稳定敲除circKIFA的HCC-1806细胞和对照细胞构建TNBC异种移植模型,研究circKIF4A在体内是否具有相同的作用。注射肿瘤细胞后,每周一次测量小鼠移植肿瘤的大小,绘制生长曲线。4周后,对小鼠进行体外荧光成像,安乐死后切除肿瘤。结果显示,与对照组相比,稳定敲低circKIF4A组移植肿瘤的大小明显减小(图2A-C)。sh-circKIF4A组肿瘤重量明显降低(图2D)。此外,对裸鼠肿瘤进行免疫组化,结果显示sh-NC组细胞增殖标志物Ki-67显著升高(图2E)。为了进一步研究circKIF4A在TNBC脑转移中的作用,我们通过左心室注射稳定敲除circKIF4A并表达荧光素酶的HCC1806细胞以及对照细胞,构建了脑转移模型(图2F)。培养50天后,再次对小鼠进行体外荧光成像,结果显示,敲低circKIF4A可显著抑制HCC1806细胞体内脑转移能力(图2G)。稳定敲低circKIF4A组脑转移灶的大小和数量明显低于对照组。circKIF4A敲低组脑转移结节数为2±1个,对照组为9±3个(图2H和图2I)。这些结果证实敲低circKIF4A可以抑制三阴性乳腺癌细胞的脑转移能力。
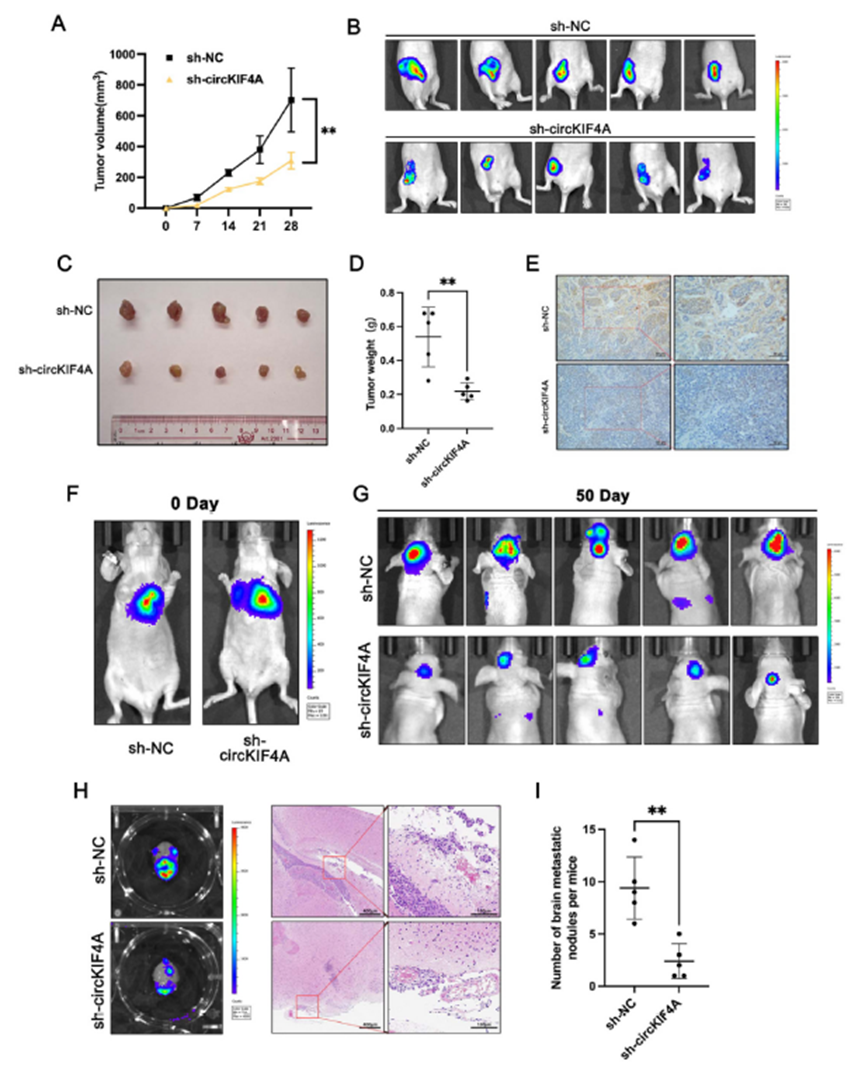
3)circKIF4A作为海绵与miR-637结合
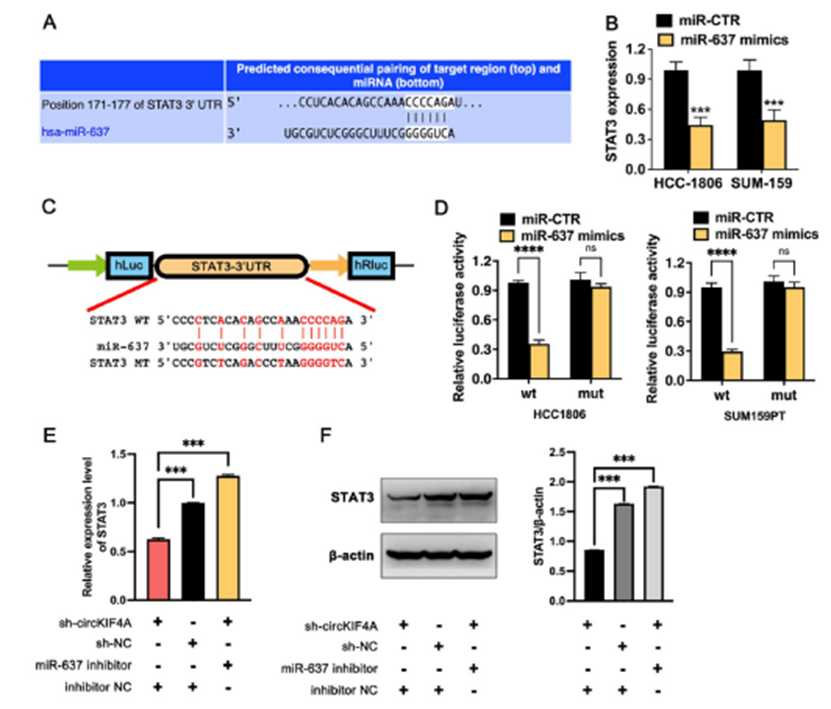
利用核细胞分离试剂盒和FISH检测circKIF4A在细胞内的分布和定位。circKIF4A主要分布在细胞质中(图3A和B)。根据Targetscan预测,circKIF4A具有miR-637结合位点(图3C)。同时,对22对三阴性乳腺癌脑转移灶和原发灶的qRT-PCR检测显示,miR-637在脑转移组织中的表达(1.84±1.16)明显低于原发灶的肿瘤组织(5.14±1.54)(图3D)。RT-qPCR检测了miR-637在TNBC细胞系中的分布,结果显示,与MCF 10A相比,miR-637在TNBC细胞系中的含量明显较低(图3D)。为了进一步验证circKIF4A与miR-637在三阴性乳腺癌细胞中的结合,我们基于circKIF4A与miR-637的潜在结合序列构建了野生型双荧光素酶报告载体和结合序列突变载体(图3F)。通过双荧光素酶报告基因实验进一步证实了TNBC细胞中circKIF4A和miR-637之间的相互作用。与转染circKIF4A-wt和miR-CTR的细胞相比,转染circKIF4A-wt和miR-637模拟物的细胞荧光素酶活性显著降低。相比之下,在转染circKIF4A-mut的细胞中,分别转染miR-CTR和miR-637模拟物后,荧光素酶活性没有差异(图3G)。这些结果表明circKIF4A在TNBC中作为海绵与miR-637结合。
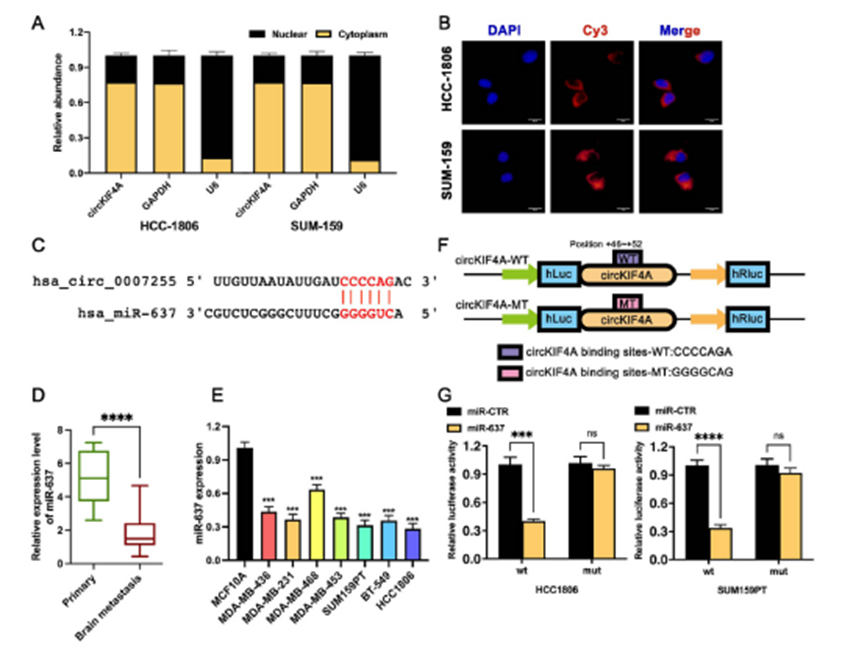
4)circKIFA通过miR-637调控STAT3的表达
通过TargetScanHuman,我们发现STAT3是miR-637的候选下游蛋白,在多种肿瘤中发挥促癌作用(图4A)。在TNBC细胞中转染miR-637模拟物后,与对照组(miR-CTR)相比,miR-637模拟物在HCC1806和SUM159PT细胞系中分别下调了54.67%±6.46和49.60%±8.50的STAT3 mRNA表达(P < 0.05)(图4B)。为了进一步验证三阴性乳腺癌细胞中miR-637是否通过结合STAT3’UTR调控STAT3的翻译,我们基于miR-637与STAT 3’UTR的潜在结合序列构建了野生型双荧光素酶报告载体和结合序列突变载体(图4C)。与转染STAT3-wt和miR-CTR的细胞相比,转染STAT3-wt和miR-637模拟物的细胞荧光素酶活性显著降低。相比之下,在转染STAT3-mut的细胞中,分别转染miR-CTR和miR-637模拟物后,荧光素酶活性没有差异(图4D)。这些结果表明miR-637通过结合STAT3 3’ UTR发挥其功能。为了验证circKIF4A是否通过circKIF4A/miR-637轴调控STAT3的表达,我们通过稳定敲低HCC1806细胞中的circKIF4A并转染相应的抑制剂,建立了敲低对照组(sh-circKIF4A + inhibitor NC)和拯救组(sh-circKIF4A + miR-637 inhibitor)。将未处理的细胞作为空白对照组(sh-NC +抑制剂NC)。采用qRT-PCR和Western blot检测上述各组中STAT3的表达变化。与空白对照组相比,敲低对照组STAT3 mRNA和蛋白的表达水平均下调;而与敲低对照组相比,拯救组STAT3 mRNA和蛋白的表达水平均有所升高(图4E和4F)。这些结果表明,circKIF4A通过结合miR-637来缓解miR-637对STAT3 mRNA的抑制作用,促进STAT3蛋白的上调,建立了circKIF4A/miR-637/STAT3调控轴。
5)STAT3过表达通过自噬恢复TNBC中circKIF4A敲低诱导的抑制作用
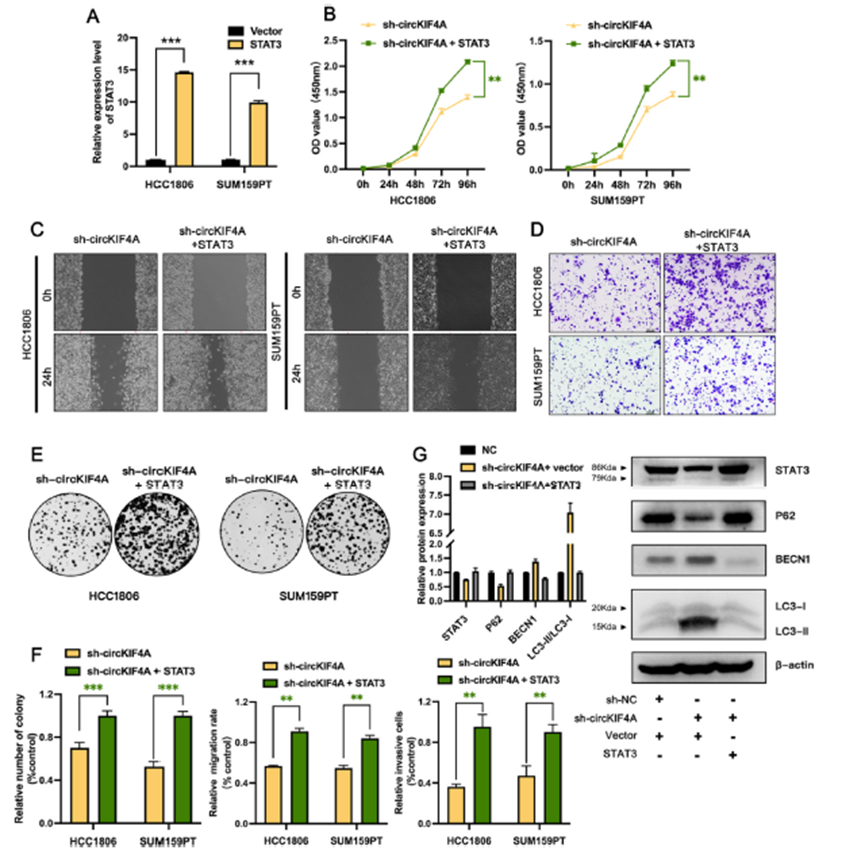
鉴于上述结果,我们开展拯救实验,探讨circKIF4A/miR-637/STAT3轴对三阴性乳腺癌细胞生物学特性的影响。我们构建了STAT3过表达载体,并利用qRT-PCR检测了STAT3在circKIF4A稳定敲低HCC1806和SUM159PT细胞系中的过表达效率。结果显示,用过表达载体转染后,STAT3的表达水平显著增加(图5A)。CCK-8和集落形成拯救实验结果表明,STAT3过表达可显著恢复TNBC的增殖能力(图5B, E, F)。Transwell和伤口愈合实验表明,STAT3过表达可以恢复TNBC细胞中被circKIF4A敲低抑制的侵袭能力(图5C, D, F)。Western blot检测证实STAT3和p62下调;但Beclin表达和LC3B-II/ LC3B-I通过敲低circKIF4A而上调,而与STAT3过表达载体共转染显著逆转了这一变化(图5D),表明抑制circKIF4A通过与STAT3竞争结合miR-637来促进自噬。这些结果可能解释了circKIF4A通过circKIF4A /miR-637/STAT3轴调控自噬进而促进TNBC脑转移的机制。
结论:
circKIF4A-miR-637-STAT3轴通过竞争性ceRNA机制调节TNBC脑转移。因此,circKIF4A是诊断和治疗TNBC脑转移的一个有前景的生物标志物。
实验方法:
qRT-PCR,CCK-8,Transwell,伤口愈合试验,集落形成试验,荧光素酶报告试验,Western blot,IHC,动物实验。
参考文献:
Wu S, Lu J, Zhu H, Wu F, Mo Y, Xie L, Song C, Liu L, Xie X, Li Y, Lin H, Tang H. A novel axis of circKIF4A-miR-637-STAT3 promotes brain metastasis in triple-negative breast cancer. Cancer Lett. 2024 Jan 28;581:216508. doi: 10.1016/j.canlet.2023.216508.