






METTL1 通过前列腺癌中 tRNA 衍生的片段生物发生促进肿瘤发生
前列腺癌(Prostate cancer, PCa)是全球第二大最常诊断的癌症,也是男性癌症相关死亡的第二大原因[1]。常规疗法针对疾病的雄激素相关信号通路。然而,高达30%的患者最终对治疗和转移产生耐药性,其治疗选择有限。随着最近大规模肿瘤样本平行测序的出现,分子分析工作揭示了高度多样化的基因组、表观基因组和转录组学景观,凸显了识别具有治疗潜力的替代改变和靶向分子通路的必要性。RNA 修饰在转移 RNA (tRNA) 中普遍存在。目前,越来越多的证据表明,tRNA及其修饰酶的失调也与肿瘤发生有关。N7-甲基鸟苷(M7G)是真核生物中最普遍的tRNA修饰之一,存在于几种tRNA物种的可变环区。在人类中,m7G由甲基转移酶1(Methyltransferase1,METTL1)和WDR4形成的复合物催化。 从功能上讲,m7G修饰的tRNA选择性地调节特定转录本的翻译。 在病理水平上,METTL1表达增加与几种癌症类型的肿瘤侵袭性有关。这些观察结果表明了tRNA修饰在癌症发展中的关键功能,并表明靶向癌症中异常的转录后修饰可能有望成为有效的治疗靶点。该文章于2023年7月发表在《Molecular Cancer》,IF:37.3
技术路线: 
研究结果:
1.METTL1 在人和PCa小鼠 中升高
为了研究RNA修饰在PCa肿瘤发生中的潜在作用,我们采用了一种确保选择与PCa肿瘤发生相关的RNA修饰的方法。我们在五项PCa研究的数据集中鉴定132组带注释的RNAmodifying proteins (RMPs)的表达变化(图1A)。此外,我们使用基因工程PCa小鼠模型将分析扩展到小鼠PCa(图1A)。我们发现原发性和转移性PCa中表达差异最大的基因是METTL1(图1B;)。从原发性到转移性人类肿瘤,METTL1表达持续增加(图1C),分析发现METTL1高表达的预后较差(图1D)。我们还发现m7g RNA甲基转移酶复合物的调控亚基WDR4的表达升高(图1C)[30,31],在其他癌症中也过表达,然而,我们没有发现WDR4过表达是不良预后的危险因素(图1D)。病人样本证实了METTL1和WDR4蛋白表达的增加(图1E)。基于前列腺癌肿瘤的激素依赖性,我们进行了METTL1、WDR4和AR的表达分析,但METTL1、WDR4与AR的表达没有明显的相关性(图1E)。为了证实METTL1和WDR4的表达是否与晚期肿瘤状态相关,我们通过S6K的磷酸化状态来测量PI3K通路的活性,在大约70%的晚期PCa患者中,S6K的磷酸化状态发生了改变。我们发现METTL1和WDR4表达与PI3K通路激活增强呈正相关(图1E),表明METTL1和WDR4表达在晚期PCa肿瘤中升高
综上所述,我们的结果表明,METTL1是PCa中改变的主要表观转录组调节因子,其过表达与不良预后相关。
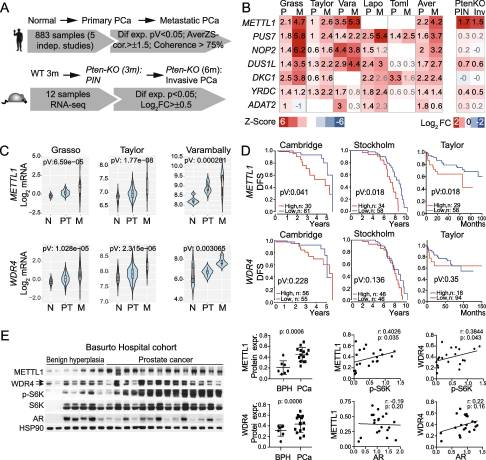
图1. METTL1 在人和PCa小鼠 中升高
2.METTL1 表达受 AKT-mTOR 下游信号通路调控
接下来,我们试图阐明前列腺癌中METTL1表达上调的机制。由于雄激素受体(AR)活性的增加是PCa的主要驱动因素之一,我们分析了通过受体和下游靶基因(如KLK3)的表达来测量的AR表达或活性的增加是否与PCa中METTL1的表达相关。在Grasso数据集中,我们观察到METTL1与AR和KLK3之间几乎存在显著的直接相关性。在Taylor数据集中,我们发现了METTL1和AR之间的直接相关性,支持了这两个因素之间潜在关系的概念。然而,在KLK3的情况下,我们没有观察到显著的相关性。我们使用TGCA数据集的分析揭示了METTL1和KLK3之间的直接相关性。然而,有趣的是,在这个特定的数据集中,我们没有发现METTL1和AR之间的显著相关性。由于我们发现与良性前列腺增生标本相比,前列腺癌标本中METTL1蛋白表达与磷酸化- s6k呈正相关(图1E),我们接下来分析了METTL1表达是否与PTEN表达相关,PTEN是PI3K/AKT/mTOR通路的负调节因子,在大约70%的晚期前列腺癌患者中缺失[47]。分析的所有数据集均显示METTL1与PTEN表达呈显著负相关(图2A),表明PI3K-mTOR轴调控了PCa中METTL1的表达。使用PI3K (BKM-120抑制剂)、AKT (MK2206)、mTORC1(雷帕霉素)和mTORC1/2 (Torin)的小分子抑制剂进一步解剖PI3K - mTORC1/2通路发现,AKT抑制降低了METTL1 mRNA的水平,mTOR抑制剂持续降低了METTL1 mRNA和蛋白的表达(图2B-C),表明METTL1的表达是通过mTOR信号传导调节的。接下来,我们研究了METTL1表达水平是否有助于确定pten缺失相关的患者生存率下降,这是具有临床意义的,并在临床局限性肿瘤患者中区分惰性和侵袭性疾病。有趣的是,我们发现高METTL1表达突出了pten -低患者预后不良的子集(图2D)。为了确定前列腺癌中METTL1的上调是否是PTEN表达低或缺失的直接后果,我们分析了野生型(WT)小鼠和Probasine-Cre x Ptenflox/flox小鼠(以下简称PTEN - ko)前列腺组织中METTL1 mRNA和蛋白水平,这些小鼠在前列腺上皮中有条件地缺失PTEN。这些小鼠在12周后发生高级别癌前病变,在5个月大后发展为浸润性腺癌。我们观察到Pten缺失后Mettl1的表达逐渐增加(图2E-G)。有趣的是,来自小鼠前列腺的trna肿瘤组织和尿液中提取的tRNA中m7g的沉积明显增加(图2H-K)。免疫组织化学分析进一步显示,Mettl1在小鼠PCa中的表达更高,在最常见的上皮细胞类型管腔细胞中也有高表达,被普遍认为是人类前列腺癌的首选起源细胞(图2L)。综上所述,这些数据表明METTL1是mTORC1通路的下游效应物,其激活可诱导METTL1在PCa中的表达增加。
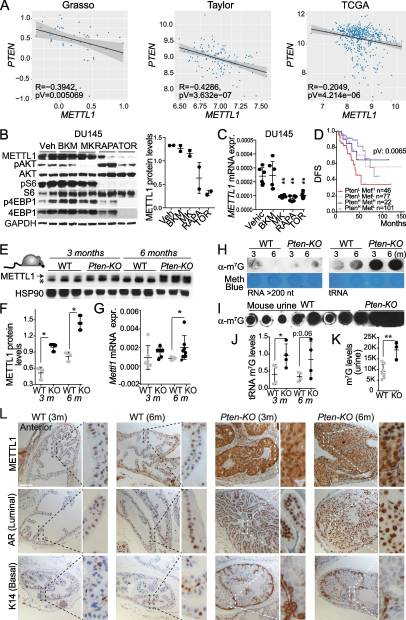
图2. METTL1 表达受 AKT-mTOR 下游信号通路调控
3.METTL1 介导tRNA甲基化
为了了解METTL1在PCa肿瘤发生中的作用,我们通过结合两种转录组范围的方法确定了METTL1 RNA底物。为了鉴定PCa细胞中mettl1特异性RNA靶点,我们使用了光活化核糖核苷增强交联免疫沉淀(PAR-CLIP),这是一种严格的技术,将光反应性核糖核苷类似物纳入新生RNA中,通过紫外线交联诱导蛋白质和RNA之间的共价键,然后进行下一代测序。以空载体感染多西环素诱导的细胞为对照。我们发现,与对照样本结合的RNA相比,t与对照样品相比,我们没有观察到HA-METTL1样品上结合的其他RNA物种的富集(图3A)。接下来,为了精确定位trna中的m7g,我们使用CRISPR/Cas9敲除PCa细胞系PC3中的METTL1(图3B)。使用抗m7g抗体的North-dot blot分析和质谱分析证实了METTL1 KO细胞trna中m7g的缺失(图3B-C)。高通量测序数据的分析证实了先前报道的鸟苷46可变环的强大甲基化(图3D)。我们鉴定出大约50%的同型受体为METTL1底物(图3E, G)。综上所述,我们的分析证实了METTL1优先甲基化PCa细胞中可变环上的trna。
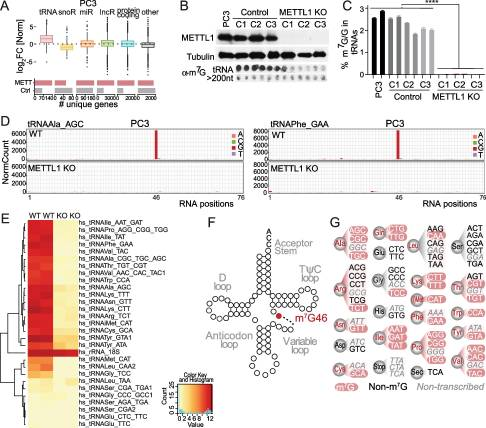
图3. METTL1 介导tRNA甲基化
4.METTL1介导的甲基化可保护tRNA免于裂解成小的非编码RNA
在酵母和人类中,当m7g甲基化降低时,trna的稳定性会降低。为了确定METTL1缺失可能会扰乱PCa细胞中单个METTL1靶向trna的水平,我们对从PC3 WT和METTL1 KO细胞中分离的trna进行了高通量测序。与之前的研究结果相反,我们没有发现证据表明mettl1特异性甲基化的缺失会降低特异性成熟tRNA同受体的丰度(图4A)。最近的报道表明,tRNA修饰保护或诱导tRNA切割成抑制性小ncrna。我们分析了来自PC3 WT和METTL1 KO细胞的小RNA测序数据,以寻找tRNA片段(trf)的主要类别的差异,包括5 ' /3 '衍生的trf (5 ' trf和3 ' trf)、5 ' tRNA一半和内部trf (int-tRF)。有趣的是,我们在PC3 METTL1 KO细胞中检测到一致的5’trna片段富集(图4B)。然而,与WT细胞相比,大多数METTL1 KO细胞的富集程度适度增加(图4C)。我们的分析显示,与WT细胞相比,在所有5'tRNA片段中,一个特定的类别,即5 '末端低鸟嘌呤tRNA片段(5'TOGs),在KO细胞中显着过度代表(log2 FC>2, p值<0.05)(图4B和C)。5 ' togs长约20或30个核苷酸,主要来源于mettl1靶tRNA Cys(产生含有5 ' togs的5个末端鸟嘌呤(5G))和Ala(产生含有5 ' togs的4G)的裂解(图4D和E)。Northern blotting证实,与WT细胞相比,PC3 METTL1 KO细胞的两个独立克隆中cys衍生的5 ' trf的积累较弱,但明显较高(图4F)。在PC3、DU145和22Rv1细胞中,METTL1下调时观察到5'tRFs,表明5'tRFs的形成与PTEN、p53和AR状态无关(图4F)。证据表明,一小部分tRNA切割成tRNA衍生的ncrna是对应激的保守反应,tRNA修饰保护它们免受应激诱导的切割。在应激反应中,tRNA切割在METTL1 KO细胞中比在WT细胞中更为突出,早在氧化应激暴露2小时达到峰值,在应激刺激8小时后下降,可能是因为无法解决应激导致细胞死亡增加(图4G, H)。总之,我们的数据表明,mettl1介导的甲基化是一种保守的机制,它调节了PCa细胞在应激反应中源自5’trna片段的一类新型小ncrna的生物发生,而不管它们的遗传状态如何。
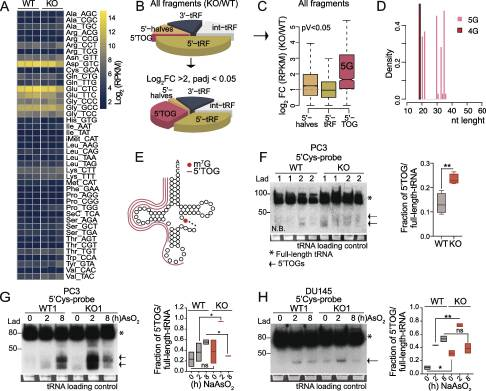
图4. METTL1介导的甲基化可保护tRNA免于裂解成小的非编码RNA
5.METTL1 的缺失通过 tRNA 片段生物发生抑制翻译起始
由于已知5'TOGs抑制全局翻译,我们接下来研究了5'TOGs的积累是否导致了PC3 mettl1缺失细胞的翻译改变。我们证实,与WT细胞相比,METTL1 KO细胞中通过o -丙基嘌呤霉素(OP-puro)掺入测量的蛋白质翻译减少(图5A)。蛋白质合成速率的变化与翻译起始复合物组分的表达变化或eIF2α的磷酸化状态无关。细胞应激反应中5’trna片段的产生在应激反应机制中起关键作用。这一过程有助于抑制整体蛋白质合成,使细胞能够恢复并在应激条件下存活[66]。为了确定METTL1的去除是否会影响应激反应,我们测量了氧化应激诱导后的蛋白质合成速率。我们的分析表明,与WT细胞相比,METTL1缺陷细胞的蛋白质合成回收率降低,这表明METTL1 KO细胞对应激更敏感(Supplementary Fig. S5B)。这一发现表明m7g的缺失显著抑制了蛋白合成,并且这一过程与eIF2α磷酸化状态无关。接下来,我们试图确定METTL1 KO细胞独特的翻译抑制及其对5'TOGs的依赖的分子基础。先前的证据表明,5'TOGs通过取代mrna帽上的翻译起始复合物eIF4A/G/E和调控因子YB1和PABP1的组分而损害翻译起始。为了确定翻译起始因子是否从PC3 METTL1 KO细胞中7-甲基-鸟苷化(m7g)覆盖的mrna中转移,我们分析了翻译起始因子对m7g -帽包被的sepharose beads的亲和力。我们发现,与WT细胞相比,METTL1 KO细胞中eIF4G和PABP1的m7 g -cap亲和力显著降低(图5B)。这表明,5'TOGs积累的增加破坏了翻译起始复合物某些因子的组装和结合的稳定性,导致METTL1 KO细胞中的翻译受到抑制。我们进一步评估了在WT细胞中表达的合成5'TOG是否能够结合翻译起始因子和调节因子,以及它们对这些因子的亲和力是否可以通过合成的逆补体5'TOG rna(或抗tog)来阻断。为此,我们用5 '生物素化的5'TOG rna(包含PC3 METTL1 KO细胞中最丰富的5'TOG序列)转染PC3 WT细胞,并添加或不添加抗tog。在去除5 '生物素化-5 ' togs后,我们发现在抗togs存在下,PABP1和YB1对5'TOG的亲和力显著降低(图5C)。接下来,我们研究了人工合成的5'TOGs是否可以在WT细胞中表型化翻译起始复合物的位移,以及抗tog rna是否可以在METTL1 KO细胞中挽救这种观察到的效应。我们发现,在转染5'TOGS的WT细胞中,PAPB1与m7 g -cap的结合被取代,但在转染抗tog rna的METTL1 KO细胞中,PAPB1与m7 g -cap的亲和力显著增加(图5D)。总的来说,这表明5'TOGs的形成通过取代PAPB1来抑制蛋白质翻译。有趣的是,PAPB1先前已被证明对其他细胞类型中的5'TOGs具有很强的亲和力。因此,我们的研究结果证实,METTL1 KO细胞中表达的5'TOGs可以取代mRNA帽上的翻译调节因子,并为METTL1介导的翻译抑制的分子基础提供了关键见解。
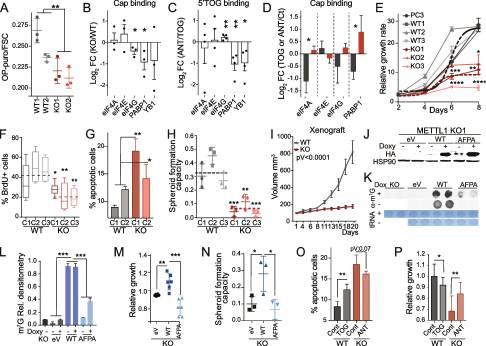
图5. METTL1 的缺失通过 tRNA 片段生物发生抑制翻译起始
5.METTL1下调可抑制前列腺肿瘤在体内和体外的生长
鉴于METTL1 KO细胞合成的蛋白质较少,我们假设METTL1抑制可以降低细胞和肿瘤的生长。事实上,我们观察到METTL1的敲低会损害PC3、DU145和22Rv1细胞的生长(图5E)。METTL1下调还会减少细胞分裂,诱导细胞周期阻滞,增加细胞凋亡,损害球体形成能力,并显著降低肿瘤异种移植物的生长和增殖(图5f - I)。因此,我们的数据表明,下调METTL1可以有效地抑制肿瘤生长,有力地支持METTL1在调节PCa进展中起关键作用。接下来,我们研究了METTL1甲基化酶活性是否足以促进细胞生长。我们在PC3 METTL1 KO细胞中重新表达了野生型METTL1 (WT)和催化死亡突变体(AFPA)(图5J-L)。与用空载体(eV)转导的KO细胞相比,重新表达WT METTL1的KO细胞的增殖和球体形成能力增强。相比之下,无催化活性突变体的表达未能促进METTL1 KO细胞生长和球体形成能力(图5M-N)。由于METTL1的催化活性得到了与调控亚基WDR4形成复合物的支持,并且WDR4在PCa中过表达(图1C, E),综上所述,这些结果表明METTL1以酶活性依赖的方式促进PCa细胞的生长,但不依赖于WDR4。为了进一步确定5'TOG是否足以诱导生长停滞和凋亡,我们用5'TOG转染PC3 WT细胞,用抗tog转染METTL1 KO细胞,并测量细胞凋亡和增殖。我们检测到WT细胞转染5’tog rna后,凋亡显著增加,增殖显著减少,而METTL1 KO细胞转染antiTOGs后,凋亡虽不显著减少,但增殖显著增加(图5O,P)。总之,我们的数据表明,METTL1失活和5'TOGs是诱导生长停滞所必需的,重新表达催化活性版本的METTL1和抗togs可以部分修复METTL1 KO细胞中观察到的缺陷。
6.METTL1 缺失激活 IFN 信号通路
鉴于翻译受到METTL1抑制的影响,我们探讨了METTL1缺失对翻译体即刻变化的影响。为此,我们利用OP-puro特性来标记新生蛋白,随后将其偶联到生物素包被的微球上,然后进行微球上的消化和LC-MS /MS(图6A)。基因本体(GO)术语富集分析发现,与细胞分裂和有丝分裂相关的基因翻译减少,证实了观察到的METTL1 KO细胞增殖缺陷(图6)。出乎意料的是,我们还发现METTL1 KO细胞中与应激反应相关的转录本翻译增加,包括I型和II型干扰素(IFN)信号通路、免疫效应过程和分解代谢过程(图6B)。翻译的差异反映在PC3 METTL1 KO细胞的整体蛋白质组组成中,但与RNA表达水平无关,这表明转录后或翻译调控是导致METTL1 KO细胞中不相关的RNA -蛋白表达水平的原因(图6C)。为了进一步测试METTL1 KO细胞中某些转录本的翻译效率是否存在差异,我们进行了多体分析。这种方法使我们能够确认METTL1 KO细胞中活性多体的形成减少,同时整体蛋白合成减少(图6D)。对METTL1 KO细胞多体部分mRNA富集的分析显示,与干扰素信号相关的特定转录物的翻译增加,如干扰素调节因子9 (IRF9)和干扰素刺激基因15 (ISG15)(图6D)。作为翻译变化特异性的对照,我们没有发现在METTL1 KO细胞的多体部分中富集GAPDH、Catenin β、KIF20A或STAT1等转录本(图6D)。综上所述,我们的研究结果表明,mettl1介导的tRNA甲基化引导着一个独特的翻译程序。由于5'TOGs可以重新编程翻译机制,以支持癌细胞所需的翻译程序,我们接下来研究了METTL1 KO细胞中的翻译变化是否由5'TOGs的生物发生增加介导。为此,我们用5'TOG和抗tog rna转导PC3 WT和METTL1 KO细胞,并评估蛋白质表达的变化。我们发现,WT细胞中转染5'TOGs增加了IRF9和ISG15蛋白的表达,而在METTL1 KO细胞中转染抗tog rna轻微但显著地降低了这两种蛋白的表达(图6E)。我们还观察到METTL1 KO细胞和异种移植物中STAT1的蛋白表达和磷酸化增加(图6E),但其表达的增加与翻译的增加无关(图6D),而是转录依赖的,这表明STAT1表达的增加是由METTL1 KO细胞中IFN信号通路的激活诱导的。事实上,其他干扰素刺激基因(ISGs)的转录也在METTL1敲除细胞中被转录激活。综上所述,我们的数据表明,mettl1介导的tRNA甲基化和tRNA片段生物发生诱导了激活IFN信号通路的翻译程序。为了证实METTL1的高表达与人类PCa样本中IFN通路活性的降低相关。
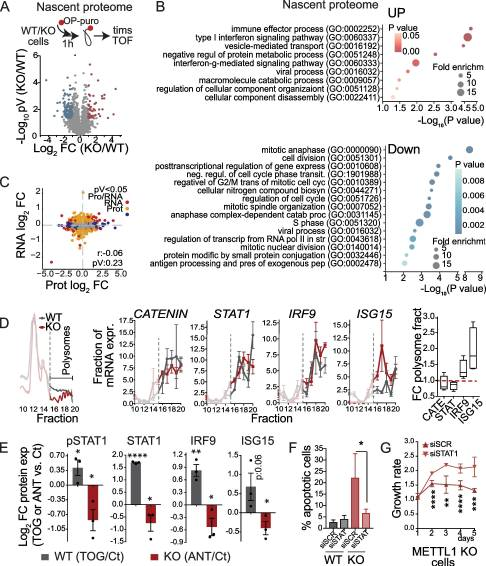
图6.METTL1 缺失激活 IFN 信号通路
7.PCa 中 METTL1 表达低与促炎免疫细胞极化增加相关
最近,表观遗传靶向治疗已被证明可以在几种癌症类型中触发IFN抗病毒反应,包括PCa,引发先天免疫反应并导致许多细胞因子的产生[74-77]。为了阐明METTL1抑制是否可以通过激活IFN信号通路引发先天免疫反应,我们研究了METTL1下调是否会改变PCa细胞中细胞因子的表达和分泌。总体而言,在METTL1 KO细胞中,细胞因子组成分析显示,参与促炎活性的细胞因子分泌增加,并使巨噬细胞极化为m1样内型[78],包括粒细胞-巨噬细胞集落刺激因子(GM-CSF)和肿瘤坏死因子α (TNF-α)(图7A)。METTL1的去除也诱导了抗炎细胞因子的下调,包括巨噬细胞或集落刺激因子(M-CSF)、IL10和IL13(图7A),可使巨噬细胞极化为m2样内型。这些数据表明,肿瘤细胞中METTL1的抑制可以使肿瘤微环境(TME)中的免疫细胞向细胞毒性杀瘤内型分化。我们用PC3 WT或METTL1 KO细胞的条件培养基(cm)培养人单核细胞系THP1,并研究了m1样和m2样巨噬细胞内源性标记物的表达(图7B)。此外,PC3 METTL1 KO细胞cm存在下培养的外周血巨噬细胞对CD3+T细胞的增殖和迁移具有更高的诱导作用(图7C, D)。总之,我们的数据表明,METTL1在癌细胞中的抑制可能会刺激TME的细胞毒性和抗肿瘤炎症反应。为了在体内验证我们的发现,我们使用免疫组织化学分析评估了人类前列腺肿瘤的肿瘤内免疫细胞组成。分析表明,在PCa石蜡样品中,m2样巨噬细胞浸润与METTL1表达之间存在显著的直接相关,但m1样巨噬细胞浸润呈相反趋势。同样,CD8+T细胞浸润与METTL1表达呈负相关(图7E)。Pten-KO小鼠前列腺上皮中Mettl1杂合缺失(Pten-KO/Mettl1+/-)和Mettl1条件缺失(Pten-KO/ mett1flox /flox)导致腹侧和前叶肿瘤体积显著减少,Pten-KO/Mettl1+/+小鼠的肿瘤体积始终较大(图7F)。基于这些观察结果和mettl1缺陷细胞引发细胞毒性免疫反应的能力,其特征是诱导m1样巨噬细胞极化,以及体外CD8+T细胞的增殖和迁移增强(图7AD)。为了阐明Mettl1抑制对免疫肿瘤组成重编程的影响,我们分析了免疫调节分子,包括细胞因子和趋化因子,作为肿瘤中存在的免疫细胞的替代标记物。我们的研究结果揭示了细胞因子组成的显著变化,其特征是促肿瘤细胞因子的分泌减少。此外,我们观察到在Pten-KO/ mettt1fl /fl肿瘤中,已知与免疫检查点阻断(ICB)治疗有利应答相关的细胞因子分泌增加(图7H)。这些发现表明,抑制Mettl1可能有助于改善ICB治疗结果的免疫微环境,突出了靶向Mettl1作为增强抗肿瘤免疫反应的治疗策略的潜力。为了确定mett1缺陷肿瘤的肿瘤内细胞因子组成是否可以增强ICB治疗的疗效,我们用抗pd1和抗ctl4a抗体治疗小鼠。与未治疗的小鼠相比,Pten-KO/Mettl1+/+小鼠在ICB治疗后未显示肿瘤体积减少,而Pten-KO/ mett1flox /flox小鼠在ICB治疗后肿瘤体积显著减少(图7I)。我们还研究了METTL1表达水平是否可以预测患者ICB治疗的疗效。使用ROC绘图仪平台,我们分析了抗pd1治疗的几种肿瘤类型中METTL1的表达水平。我们发现,在乳腺癌、结直肠癌、卵巢癌和胶质母细胞瘤患者中,对ICB治疗有反应的METTL1表达低于无反应的METTL1表达,这表明高METTL1表达预示着对ICB治疗的不良反应(图7J)。总之,我们的研究结果表明,METTL1的表达水平可以决定肿瘤内细胞毒性免疫浸润和ICB治疗的成功。
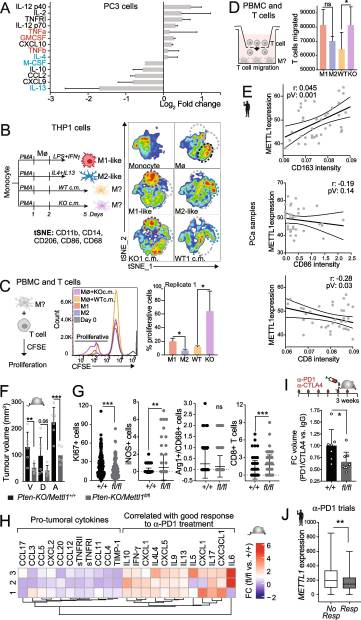
图7.PCa 中 METTL1 表达低与促炎免疫细胞极化增加相关
结论:
本研究表明,我们发现转移RNA N7 -甲基鸟苷(m7g)转移酶METTL1在原发性和晚期前列腺肿瘤中高度表达。从机制上讲,我们发现METTL1缺失导致m7 G tRNA甲基化缺失,并促进了一类来自5'tRNA片段的新型小非编码rna的生物发生。5 ' trna衍生的小rna引导翻译控制,有利于肿瘤生长抑制、干扰素途径和免疫效应器的关键调节因子的合成。在前列腺癌临床前模型中,Mettl1的敲低增加了促炎免疫细胞在肿瘤内的浸润,增强了对免疫治疗的反应。总的来说,我们的研究结果揭示了mettl1导向的m7g tRNA甲基化在癌细胞翻译控制和肿瘤生物学中的治疗作用。
实验方法:
细胞培养、稳转细胞系建立、WB、RT-qPCR、免疫组化、免疫荧光、mRNA-seq、PAR-CLIP、PAR-CLIP analysis、AlkAniline-seq、tRNA-seq、tRNA-seq analysis、Northern blotting、帽结合试验。
参考文献:
METTL1 promotes tumorigenesis through tRNA-derived fragment biogenesis in prostate cancer. Mol Cancer. 2023;22(1):119. Published 2023 Jul 29.