






长链非编码RNA Malat1可预防骨质疏松和骨转移
骨质疏松症以骨密度降低、骨脆性增加和骨折易感性增加为特征,反映了破骨细胞骨吸收超过成骨细胞骨形成的不平衡,并加速骨转移。原发性骨质疏松发生在衰老过程中,尤其是绝经后女性。继发性骨质疏松与原发性骨质疏松的结局相同,但由某些疾病或药物引起。在这两种情况下,过度的破骨细胞生成起着关键作用,并为治疗干预提供了机会。破骨细胞是一类多核巨细胞(MGC),起源于单核/巨噬细胞谱系,负责骨基质和矿物质的吸收。破骨细胞的生成由巨噬细胞集落刺激因子(M-CSF)和核因子-κB受体活化因子配体(RANKL)启动,诱导破骨细胞标志物如组织蛋白酶K(CTSK)和酸性磷酸酶5(ACP5,也称为TRAP)的表达,随后破骨细胞前体成熟和细胞-细胞融合。作为破骨细胞生成的主要调节因子,活化T细胞核因子1(NFATC1)被RANKL诱导,进而与其他转录因子形成复合体,激活其编码基因以及其他参与破骨细胞黏附、细胞融合和骨吸收的基因的转录。长链非编码RNA(lncRNA)是长度超过200个核苷酸且不翻译成蛋白质的转录本,通过与DNA、其他RNA和蛋白质结合发挥功能。lncRNA通常进化保守性较低。MALAT1是一个高度保守的核内长链非编码RNA,在的组织中大量表达。根据siRNA敲低培养细胞系的结果,MALAT1已被证明调节选择性前体mRNA剪接。然而,目前缺乏MALAT1改变在低BMD和骨质疏松中发挥作用的功能证据。该研究发表在《Nature Communications》,IF:16.6。
技术路线:

主要研究结果:
1. MALAT1在人类和小鼠破骨细胞分化过程中下调
CMPs可分化为单核细胞和巨噬细胞,是破骨细胞的前体。作者分析了人胎盘CD14+巨噬细胞向MGCs分化过程中的基因表达(图1a),其中破骨细胞是的主要细胞群。与CD14+巨噬细胞相比,MGCs中破骨细胞标志物的表达升高,包括NFATC1、CTSK、DCSTAMP、ATP6V0D2、ATP6V0E2和ATP6V0A1(图1b,c)。相反,相对于CD14+巨噬细胞,MALAT1在MGCs中显著下调(图1a-c)。与破骨细胞的功能一致,基因集富集分析表明,与CD14+巨噬细胞相比,在MGCs中富集的基因集与胶原组织、细胞外结构重塑和骨骼发育相关(图1d)。为了进一步验证Malat1在巨噬细胞向破骨细胞分化过程中的下调,作者用可溶性RANKL处理小鼠巨噬细胞/前破骨细胞细胞系RAW264.7,以诱导破骨细胞分化。在此处理后,破骨细胞的标志物,包括NFATC1,Ctsk和Trap5,以时间依赖性的方式上调(图1e-g),而Malat1的表达水平显著降低(图1h)。综上所述,这些结果揭示了MALAT1作为一个lncRNA在人类和小鼠的破骨细胞生成过程中下调。
脂多糖(LPS)和TNF-α参与病理性破骨细胞生成。与之前的报告一致,作者观察到单独的LPS处理不足以启动破骨细胞分化;相反,当RAW264.7细胞用RANKL预处理时,LPS处理促进了破骨细胞的生成,如抗酒石酸酸性磷酸酶(TRAP)染色所示(图1i,j),这是一种广泛使用的破骨细胞标志物(图1k),并上调了NFATC1和Ctsk的表达(图1k)。另一方面,TNF-α可以通过依赖和不依赖RANKL的方式诱导破骨细胞分化。事实上,作者发现用TNF-α处理RAW264.7细胞诱导的破骨细胞生成和NFATC1和Ctsk的表达,无论是否用RANKL预处理(图1i,l,m)。值得注意的是,Malat1在LPS或TNF-α诱导的破骨细胞分化过程中显著下调(图1k,m)。这些发现表明,Malat1可能参与对各种刺激做出反应的破骨细胞生成。
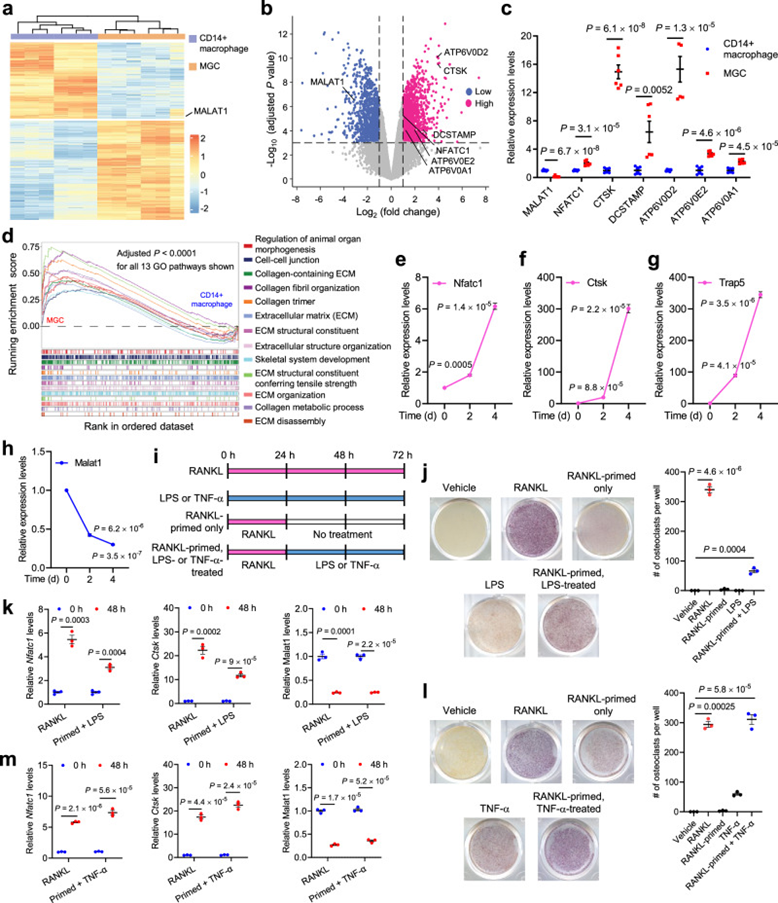
图1 MALAT1在破骨细胞分化过程中表达下调
2. 遗传模型显示Malat1可预防骨质疏松和骨转移
为了研究Malat1在破骨细胞生成和骨质疏松中的作用,作者使用了作者之前研究中描述的Malat1敲除小鼠模型(Malat1−/−),在Malat1的转录起始位点的下游插入一个转录终止子,导致Malat1 RNA表达的丢失,而不改变Malat1的邻近基因的表达水平。此外,作者之前在小鼠中构建了ROSA26基因座的Malat1转基因靶向表达(Malat1Tg/Tg),这使作者能够通过产生Malat1−/−;Malat1Tg/Tg动物,在Malat1−/−小鼠中进行遗传拯救研究。作者收集了Malat1+/+,Malat1−/−,Malat1−/−小鼠的各种组织,包括骨髓,胃,结肠,小肠,肝脏和胰腺组织;Malat1Tg/Tg小鼠,通过qPCR检测Malat1的表达水平。
通过对6个月大的动物进行股骨微计算机断层扫描(μCT)分析,作者发现雄性和雌性Malat1−/−小鼠的骨密度比年龄和性别匹配的Malat1+/+小鼠低得多;重要的是,这种骨质疏松表型在Malat1−/−;Malat1Tg/Tg小鼠中得到了挽救(图2a,b)。μCT数据的定量显示,与Malat1+/+小鼠相比,骨小梁密度(图2c,d)在Malat1−/−小鼠中显著减少,并且这些减少在很大程度上被Malat1表达的遗传恢复所逆转(图2c,d)。TRAP染色显示,与Malat1+/+小鼠相比,Malat1−/−小鼠股骨中破骨细胞显著增加,并且这种增加在Malat1−/−;Malat1Tg/Tg小鼠中被逆转(图2e)。股骨破骨细胞的定量分析显示,相对于Malat1+/+小鼠,每骨周长的破骨细胞数量显著增加(图2f)和每骨表面的破骨细胞表面(图2g)在Malat1−/−小鼠中升高约2倍。
接下来,为了确定Malat1在调节骨病理性丢失中的作用,作者使用了一个成熟的炎症性骨吸收小鼠模型,包括向颅骨皮下注射LPS。μCT成像显示,与Malat1+/+或Malat1−/−;Malat1Tg/Tg小鼠相比,给8周龄的Malat1−/−小鼠注射LPS后,颅骨表面的骨质破坏显著加重(图2h,i)。TRAP染色和定量显示,注射LPS后,Malat1−/−小鼠的每骨周长有更高的破骨细胞数量(图2j,k)和更多的破骨细胞表面每骨表面(图2j,l),Malat1+/+或Malat1−/−;Malat1Tg/Tg小鼠。综上所述,这些发现表明Malat1缺乏促进生理和炎症条件下的骨质疏松。
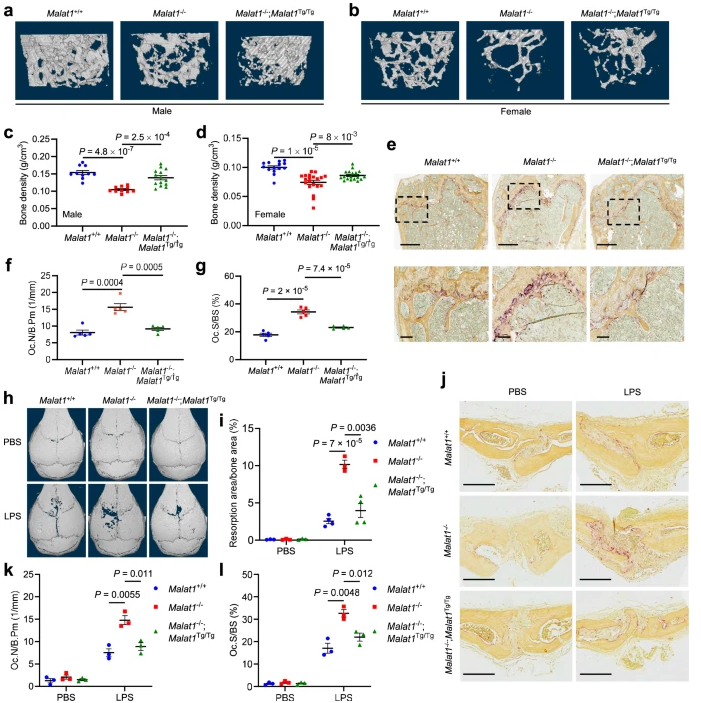
图2 Malat1预防骨质疏松
未经治疗的骨质疏松症与的癌症患者骨转移加速相关。治疗骨质疏松的药物,如抑制破骨细胞介导的骨吸收的双膦酸盐,已被用于治疗骨疾病,包括骨转移。为了确定宿主中的Malat1是否对骨转移具有保护作用,作者将荧光素酶标记的B16F1黑色素瘤细胞注射到6个月大的雄性Malat1+/+,Malat1−/−,或Malat1−/−;Malat1Tg/Tg小鼠的胫骨中,作者发现宿主中的Malat1缺失显著加剧了骨转移,Malat1的重新表达挽救了这一表型。通过活体动物的生物发光成像(图3a)和解剖的骨骼(图3b,c),以及骨骼中可见肿瘤的肉眼检查进行测量(图3d)。
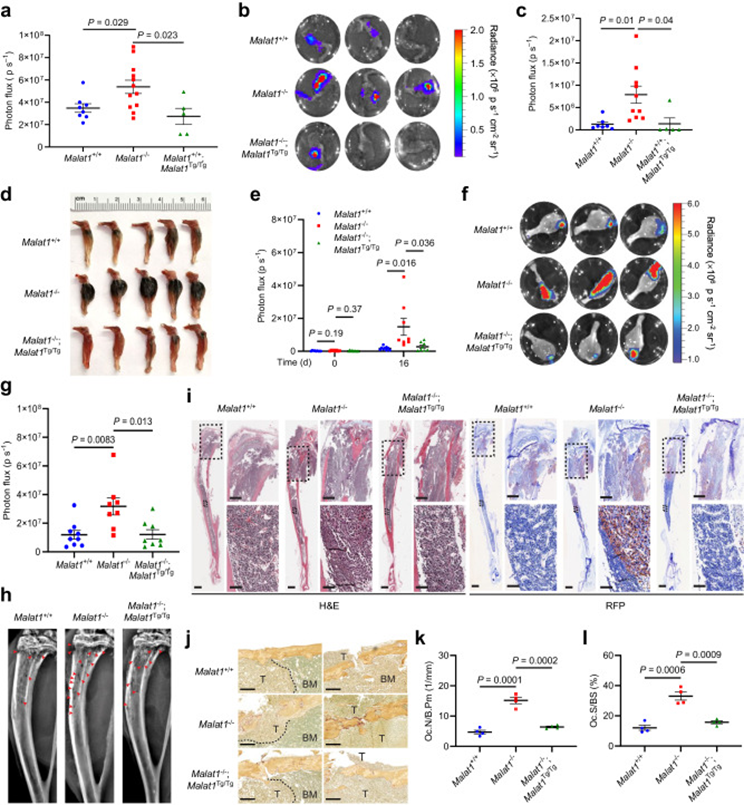
图3 Malat1保护黑色素瘤和乳腺肿瘤细胞的骨转移
鉴于骨是乳腺癌的常见转移部位,作者在3个月大的雌性Malat1+/+,Malat1−/−,和Malat1−/−;Malat1Tg/Tg小鼠的胫骨内注射EO771细胞系,EO771细胞系来源于C57BL/6背景下的小鼠乳腺肿瘤。注射2×105荧光素酶标记的EO771细胞后,3组动物在注射当天的生物发光信号基线水平无明显差异。在研究终点,作者观察到Malat1−/−小鼠的信号显著高于Malat1+/+和Malat1−/−;Malat1Tg/Tg小鼠(图3e)。安乐死后,作者收集胫骨进行离体成像(图3f,g),证实了在体成像结果。作者还对胫骨进行了x线成像,发现Malat1−/−小鼠有更多的溶骨性病变(图3h)。此外,骨切片的he染色显示,Malat1−/−小鼠的胫骨中有更高的肿瘤负荷,证据是靠近生长板的骨皮质中有更多的癌变,肿瘤区域更深地延伸到远端骨髓腔(图3i)。免疫组织化学染色RFP(与荧光素酶共表达)支持组织学分析(图3i)。此外,TRAP染色显示,与Malat1+/+和Malat1−/−;Malat1Tg/Tg小鼠相比,Malat1−/−小鼠胫骨中破骨细胞数量增加(图3j-1)。结合B16F1模型的结果,这些发现共同表明,宿主小鼠中Malat1的缺失加剧了黑色素瘤和乳腺癌细胞的转移性骨定植。
3. 骨质疏松、骨肉瘤或乳腺癌骨转移患者骨组织的单细胞转录组分析
为了评估MALAT1在骨质疏松和骨转移中的临床相关性,作者分析了人类骨组织的单细胞RNA-seq数据。数据集包括:GSE190772样本来自2例乳腺癌骨转移患者,GSE162454样本来自6例骨肉瘤患者,以及GSE169396特征为1例非骨质疏松个体和3例骨质疏松患者(髋关节置换术中采集的股骨头)的骨组织。骨肉瘤和乳腺癌骨转移常表现出溶骨性特征。
作者使用“和谐”法去除样本间的批间效应,随后应用降维法对基于标记基因的细胞类型进行注释。这些分析确定了不同患者组的细胞簇特异性转录组。然后,作者分析了非骨质疏松个体(图4a,b)、骨质疏松患者(图4c,d)、骨肉瘤患者(图4e,f)和乳腺癌骨转移患者(图4g,h)的前破骨细胞(包括单核细胞和巨噬细胞)和成熟破骨细胞中MALAT1的表达。在每组中,与前破骨细胞相比,MALAT1在破骨细胞中的表达水平显著降低(图4b,d,f,h)。骨质疏松、骨肉瘤或乳腺癌骨转移患者的前破骨细胞和破骨细胞中MALAT1的表达水平显著低于非骨质疏松个体(图4i-k)。这些发现表明,破骨细胞中MALAT1表达的降低与骨质疏松和骨病变相关,包括乳腺癌转移和骨肉瘤。
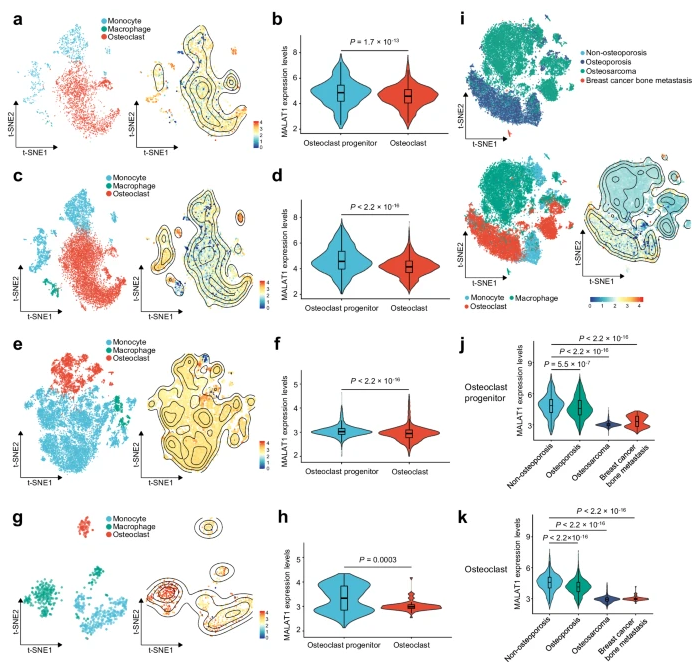
图4:骨质疏松症、骨肉瘤或乳腺癌骨转移患者骨组织的单细胞转录组分析
4. Malat1缺陷通过激活NFATC1促进破骨细胞生成
由于作者研究中使用的Malat1−/−和Malat1Tg/Tg动物是全身敲除和转基因小鼠,因此上述观察到的骨质疏松表型可能是也可能不是破骨细胞前体中Malat1缺失的直接影响。为了解决这个问题,作者从Malat1+/+,Malat1−/−,和Malat1−/−;Malat1Tg/Tg小鼠中分离出原代骨髓来源的巨噬细胞(BMMs),然后用M-CSF和RANKL处理这些破骨细胞前体4-6天,诱导其向破骨细胞分化。qPCR证实了BMMs中的基因消融和Malat1表达恢复(图5a)。在M-CSF和RANKL诱导分化后,作者通过TRAP染色检测破骨细胞,发现敲除Malat1导致TRAP阳性多核破骨细胞数量显著增加,而重新表达Malat1逆转了观察到的破骨细胞生成的诱导(图5b)。破骨细胞标志物Ctsk和Trap5的mRNA水平在Malat1−/−细胞中明显高于Malat1+/+和Malat1−/−;RANKL处理后Malat1Tg/Tg细胞(图5c,d)。
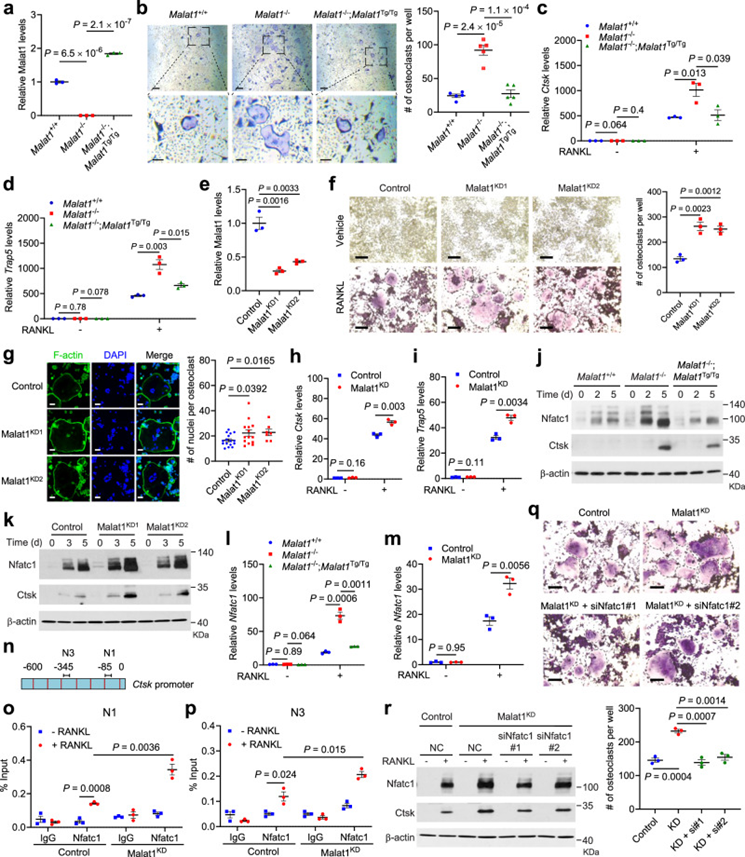
图5 Malat1缺陷通过NFATC1促进破骨细胞生成
作者还使用CRISPR干扰(CRISPRi)在细胞系中敲低Malat1。选取sgRNA-2和sgRNA-3在RAW264.7细胞中敲低Malat1,并通过qPCR进行验证(图5e),得到的两个敲低Malat1的稳定细胞株分别命名为Malat1 KD1和Malat1 KD2。RANKL诱导分化后,Malat1 KD1和Malat1 KD2细胞比对照RAW264.7细胞产生更多的trap阳性多核破骨细胞(图5f)。鬼笔环肽和DAPI分别对F-actin环和细胞核进行荧光染色,结果显示Malat1 KD1和Malat1 KD2细胞的每个破骨细胞的细胞核数量高于对照细胞(图5g)。此外,在RANKL处理的RAW264.7细胞中,敲低Malat1可以上调Ctsk和Trap5的mRNA水平(图5h,i)。总的来说,原代骨髓基质细胞和RAW264.7细胞的结果表明,破骨细胞前体中Malat1的缺失促进了RANKL诱导的破骨细胞生成。
接下来,作者将RANKL刺激时间延长至2天和5天,发现Malat1缺乏不影响Mitf、Erk1/2、c-Fos、IκBα、Creb1和p38的水平。另一方面,与Malat1+/+和Malat1−/−;Malat1Tg/Tg骨髓基质细胞相比,Malat1敲除骨髓基质细胞更多地诱导了NFATC1及其转录靶Ctsk(图5j)。作者在RANKL处理的Malat1 KD1和Malat1 KD2 RAW264.7细胞中观察到类似的结果(图5k)。已知NFATC1作为其自身的转录因子发挥作用。与Malat1野生型相比,Malat1敲除的BMMs和RAW264.7细胞在RANKL刺激后显示出更多的NFATC1 mRNA水平的诱导(图5l,m)。Ctsk,一个经典的NFATC1靶基因,包含两个NFATC1结合位点在启动子区域(图5n)。染色质免疫沉淀-qPCR检测显示,在RANKL处理后,Malat1敲低的RAW264.7细胞显示NFATC1占据了比对照RAW264.7细胞更多的这两个区域(图5o,p)。重要的是,在敲低Malat1的RAW264.7细胞中,敲低NFATC1逆转了RANKL刺激下对破骨细胞生成和Ctsk表达的诱导(图5q,r)。综上所述,这些结果表明Malat1缺失通过NFATC1促进破骨细胞分化。
作者还试图确定Malat1过表达的影响。Malat1是一种高度富集的lncRNA,作者通过多种方法进行了测试,并仅通过piggyBac转座子系统和电穿孔在RAW264.7细胞中过表达Malat1。在野生型细胞和小鼠中实现显著的Malat1过表达的挑战限制了对其过表达效应的全面研究。然而,考虑到前破骨细胞和破骨细胞中MALAT1表达的减少与骨质疏松和骨转移相关(图4i-k),作者的功能缺失方法,再加上MALAT1在MALAT1缺陷小鼠和细胞系中的重新表达,适合于这项研究。
5. Malat1结合TEAD3抑制NFATC1活性和破骨细胞生成
Malat1如何调控NFATC1与其他蛋白的结合可以激活或抑制NFATC1的转录活性,而lncRNA通常通过与蛋白的相互作用来发挥其功能,Malat1已经证明了这种作用模式。作者推测Malat1可能通过与NFATC1和/或其结合蛋白相互作用来调节NFATC1的自动扩增环路,因此作者搜索了蛋白质-蛋白质相互作用数据库Mentha(http://mentha.uniroma2.it/index.php)。
作者检测了4个Tead家族成员在BMMs和RAW264.7细胞以及其他几种小鼠细胞系中的蛋白水平。有趣的是,Tead1和Tead共激活因子Yap在BMMs和RAW264.7细胞中不可测,但在小鼠黑色素瘤细胞系B16F1、小鼠胚胎成纤维细胞、MSC和小鼠成纤维细胞系L929中大量表达(图6a)。相反,TEAD3在原发性骨髓基质细胞中表现出相对特异性的表达模式。为了确定前破骨细胞中Malat1是否与NFATC1相互作用,作者进行了RIP检测,发现Malat1在RAW264.7细胞的pan-Tead和TEAD3免疫沉淀中均富集(图6b,c),这验证了在这些破骨细胞前体中Malat1和TEAD3之间的相互作用。为了确定Malat1是否直接结合TEAD3,作者对Malat1的6个非重叠的生物素化片段,作者发现所有6个Malat1片段,而不是一个无关的核RNA U1,都与TEAD3蛋白结合(图6d),这表明TEAD3结合位点可能弥漫性分布在Malat1上。
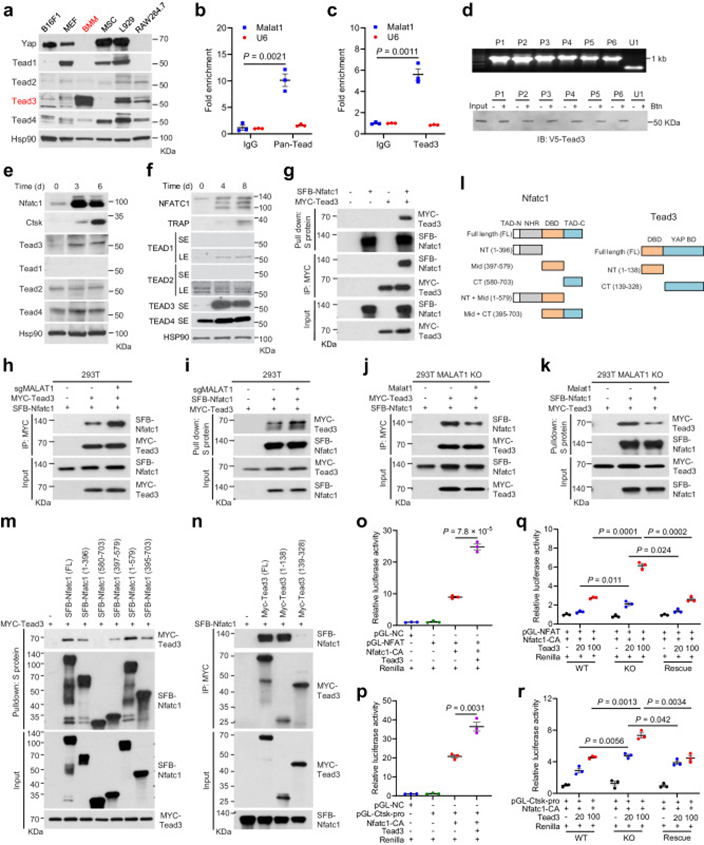
图6 Malat1与TEAD3结合抑制NFATC1活性
有趣的是,RAW264.7和U937细胞的RANKL处理上调了TEAD3,但没有上调其他Tead家族成员(图6e,f)。免疫共沉淀(co-IP)实验表明,TEAD3,而不是Yap,与NFATC1相互作用(图6g)。在验证TEAD3与Malat1和NFATC1的相互作用后,作者试图确定Malat1是否调节TEAD3与NFATC1的结合。为此,作者产生了Malat1敲除的HEK293T细胞,并将TEAD3和NFATC1转染这些细胞。Co-IP检测表明,Malat1缺失显著增加了TEAD3和NFATC1之间的相互作用(图6h,i)。为了进一步证实这一结果,作者在Malat1敲除的HEK293T细胞中重新表达Malat1,发现恢复Malat1表达降低了TEAD3-NFATC1的相互作用(图6j,k)。
NFATC1蛋白包含四个结构域:n端和c端区域的两个转录激活结构域(TAD)、一个中央DNA结合结构域(DBD)和一个n端调节结构域(图6l)。TEAD3蛋白主要由两个结构域组成:n端DBD(也称为TEA结构域)和c端yap结合结构域。因此,作者产生了NFATC1和TEAD3的截短突变体(图6l),并进行了co-IP检测,发现Nfact1的n端区域和中央DBD,而不是Nfact1的c端TAD,可以结合TEAD3(图6m)。此外,使用TEAD3截短突变体和全长NFATC1进行的co-IP分析表明,TEAD3的TEA结构域,而不是yap结合结构域,负责与NFATC1的相互作用(图6n)。
作者进一步研究了Malat1和TEAD3是否调节NFATC1的转录活性,通过使用包含串联NFATC1结合位点或Ctsk启动子的荧光素酶报告,作者发现TEAD3的过表达确实增强了NFATC1的转录活性(图6o,p)。作者发现,与野生型或Malat1挽救的细胞相比,在Malat1敲除细胞中,TEAD3过表达导致NFATC1活性出现更高的剂量依赖性增加(图6q,r)。这些结果支持以下模型:Malat1缺失解除对TEAD3的抑制,而TEAD3进而结合并激活NFATC1。
利用CRISPR激活方法激活RAW264.7细胞内源性TEAD3表达(图7a、b),作者发现过表达TEAD3促进了破骨细胞分化(图7c、d),上调了NFATC1、Trap5和Ctsk的表达(图7e-h)。接下来,作者在RAW264.7细胞中敲低TEAD3(图7i),发现敲低TEAD3减弱了RANKL诱导的破骨细胞分化(图7j,k),并下调了NFATC1和Ctsk的表达(图7l)。此外,在敲除Malat1的RAW264.7细胞中,沉默TEAD3可以抵消RANKL介导的破骨细胞生成和NFATC1和Ctsk表达的诱导(图7m-p)。因此,Malat1缺失以TEAD3和NFATC1依赖的方式促进破骨细胞分化。
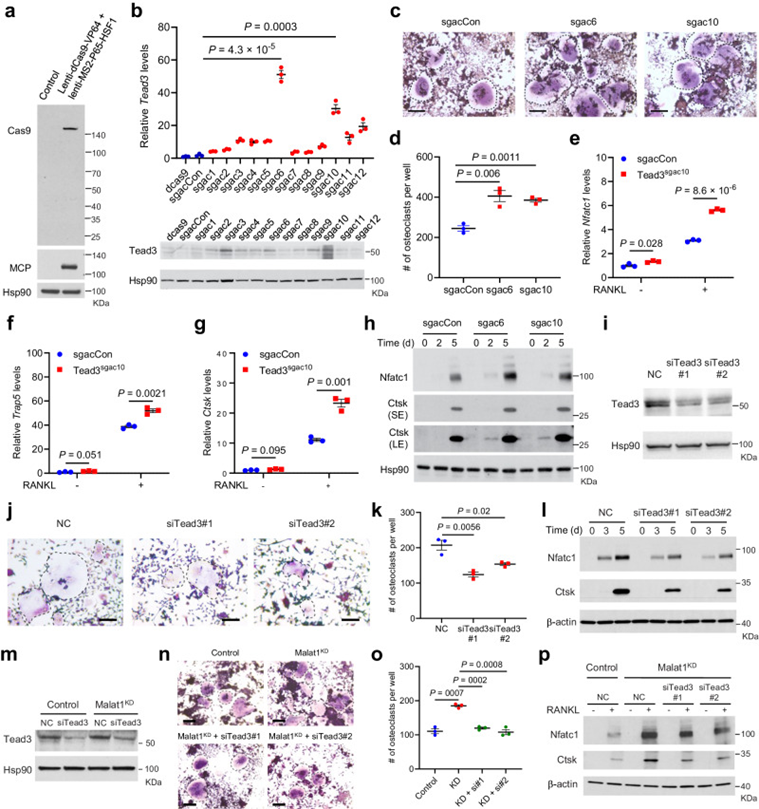
图7 TEAD3促进破骨细胞生成并介导Malat1缺陷的作用
结论:
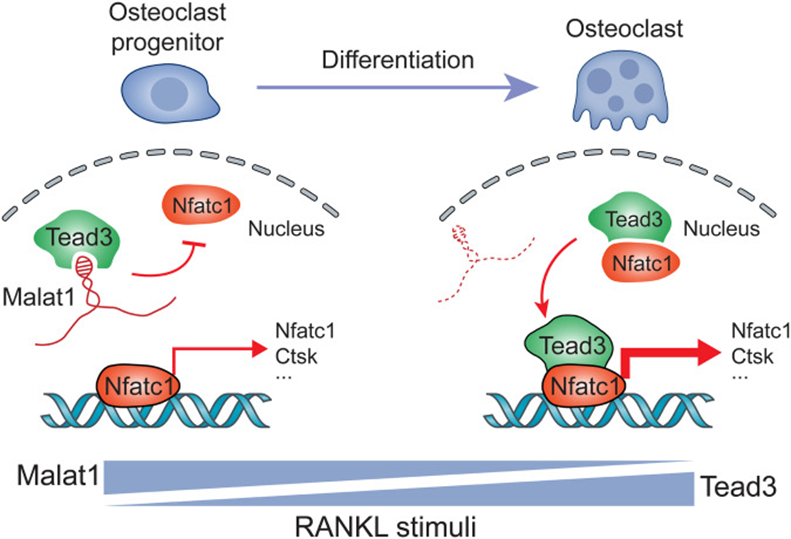
该研究发现Malat1在人类和小鼠的破骨细胞生成过程中下调。值得注意的是,小鼠中Malat1的缺失促进了骨质疏松和黑色素瘤和乳腺肿瘤细胞的骨转移,而Malat1的基因反加可以挽救这一过程。机制上,Malat1与TEAD3蛋白结合,阻断TEAD3与NFATC1的结合和激活,从而抑制NFATC1介导的基因转录和破骨细胞分化。值得注意的是,临床骨样本的单细胞转录组分析表明,前破骨细胞和破骨细胞中MALAT1表达的降低与骨质疏松和转移性骨病变相关。综上所述,这些发现证实了Malat1是一个可以预防骨质疏松和骨转移的lncRNA。
实验方法:
基因工程小鼠模型,LPS诱导的炎性骨质疏松模型,μCT骨扫描和分析,骨转移测定,骨组织组织学,TRAP染色,甲苯胺蓝染色,免疫组织化学染色,骨组织钙黄绿素染色,细胞培养,破骨细胞分化,成骨细胞分化,肌动蛋白环染色,质粒,慢病毒转导,Malat1敲低、敲除和过表达,TEAD3敲低和过表达,RNA干扰,免疫沉淀,免疫印迹,RNA提取,cDNA合成,定量PCR,RT-PCR,染色质免疫沉淀分析,RNA pulldown,RIP,荧光素酶报告基因检测,酶联免疫吸附实验,批量RNA测序分析,单细胞RNA-seq分析
参考文献:
Zhao Y, Ning J, Teng H, Deng Y, Sheldon M, Shi L, et al. Long noncoding RNA Malat1 protects against osteoporosis and bone metastasis. Nat Commun. 2024 Mar 16;15(1):2384. doi: 10.1038/s41467-024-46602-3. PMID: 38493144; PMCID: PMC10944492.