






METTL16的乳酸化通过对FDX1 mRNA进行m6A修饰在胃癌中促进铜死亡
铜死亡(cuproptosis)是由铜离子浓度过高引起的一种现象,被紧急地开发为潜在的癌症治疗方法。然而,肿瘤中铜死亡的启动、传播和最终执行机制尚不清楚。在这项研究中,作者发现在胃癌(GC)中,特别是在恶性肿瘤中,铜含量显著升高。筛选发现,METTL16,一种非典型的甲基转移酶,通过对FDX1 mRNA进行m6A修饰,在铜死亡中起着关键的调节作用。此外,铜应激促进METTL16在K229位点的乳酸化,随后引发铜死亡。METTL16的乳酸化过程受SIRT2的抑制。提高的METTL16乳酸化显著提高了铜离子载体elesclomol的治疗效果。将elesclomol与SIRT2特异性抑制剂AGK2结合使用,在体内外诱导了胃肿瘤的铜死亡。这些结果揭示了非组蛋白蛋白质METTL16乳酸化对肿瘤中铜死亡的重要性。鉴于GC中铜和乳酸浓度较高,诱导铜死亡成为一种有前景的GC治疗策略。该研究于2023年10月发表于《Nature Communications》上,题为“Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer”,影响因子16.6。
技术路线

研究方法
1. 胃癌中铜含量较高并与肿瘤进展相关
铜死亡是由过高的铜浓度引起的,但肿瘤中铜死亡的调控机制仍需进一步探讨。鉴于铜主要通过胃和上消化道吸收,因此,胃肠系统肿瘤适合研究铜死亡的启动和传播。作者分析了48对胃癌组织和相邻组织中的铜浓度,发现胃癌组织中的铜浓度高于正常胃组织(图1a,P < 0.01)。进一步分析显示,III期胃癌患者中的相对铜含量高于I期和II期胃癌患者(图1b,P < 0.05),表明铜含量与肿瘤进展相关。此外,铜含量与胃癌患者的总生存期(OS)和无病生存期(DFS)呈负相关(图1c, d; log-rank P < 0.05)。同时,铜含量与细胞增殖指标Ki-67的表达呈正相关(图1e)。同样,患者血清标本中的铜含量与血小板/淋巴细胞比值和中性粒细胞/淋巴细胞比值呈正相关,这两者都是与胃癌侵袭性恶性肿瘤相关的炎症预后生物标志(图1f, g)。这些结果表明高铜含量参与了胃癌的进展和发展。
此外,作者对不同类型的胃癌,特别是恶性肿瘤类型中铜浓度的差异进行了研究。有趣的是,作者发现粘液腺癌的铜浓度高于总体胃肿瘤(图1h)。在粘液腺癌中,铜含量与淋巴结转移(图1i)和血管侵袭(图1j)等侵袭指标呈正相关。然而,在非粘液腺癌中,铜含量与这些侵袭指标或肿瘤分化无显著相关性。粘液腺癌是胃癌的一种罕见组织学亚型,与更晚期和较差的5年总生存期和无病生存期相关39。作为难治性胃癌,粘液腺癌以化疗放疗耐药性为特征,迫切需要更有效的治疗方法。
最近,研究人员发现高胞内铜浓度会触发一种还原酶FDX1将Cu2+还原为更具毒性的Cu1+,导致铜死亡11。作者发现与正常胃组织相比,胃癌中FDX1蛋白水平较高(图1k)。考虑到胃癌中FDX1蛋白水平和铜含量都较高,铜死亡可能更容易被触发。这为胃癌提供了潜在的治疗策略,特别是对于恶性肿瘤——粘液腺癌。
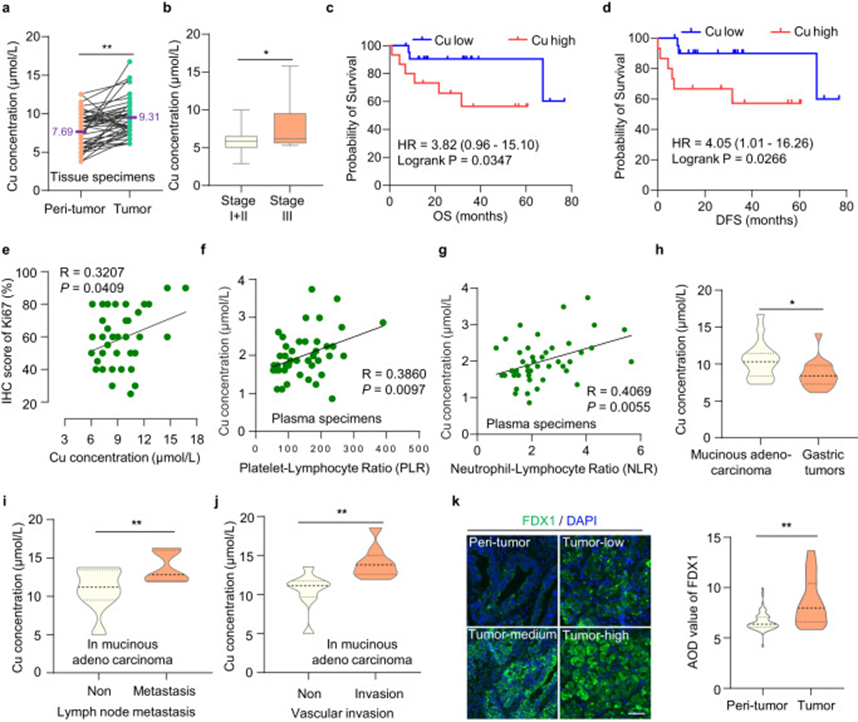
图1 胃癌中铜含量较高并与肿瘤进展相关。
2. METTL16是铜死亡的重要介导因子
已经报道与良性胃疾病和健康对照组相比,胃癌组中总m6A水平显著增加,并促进了胃癌的肿瘤发生、生长、侵袭、上皮间质转化(EMT)、转移,甚至多药耐药过程。此外,m6A修饰参与了几种细胞死亡类型,包括凋亡、坏死, 铁死亡和火死亡。然而,m6A修饰与铜死亡之间的关系仍不明确。有趣的是,作者发现胃癌组织中铜含量与总m6A水平呈正相关(R = 0.5116)。此外,铜显著上调了胃癌细胞中总m6A修饰水平。鉴于甲基转移酶样蛋白(METTL)家族在促进m6A修饰中起主导作用,作者分析了METTLs敲除细胞在以1:1比例处理elesclomol/disulfiram和铜时的存活率。结果表明,METTL16敲除导致对elesclomol/disulfiram-Cu治疗的耐药性。此外,使用硫代硫氧钼酸铜(TTM),一种铜死亡抑制剂,抑制了对照组和METTL16敲除细胞中的铜死亡,表明METTL16参与了铜死亡。为了进一步验证这些结果,成功构建并验证了METTL16敲除克隆细胞系,观察到了与METTL16敲除细胞相似的结果。这些结果表明METTL16在铜死亡中发挥着至关重要的作用。
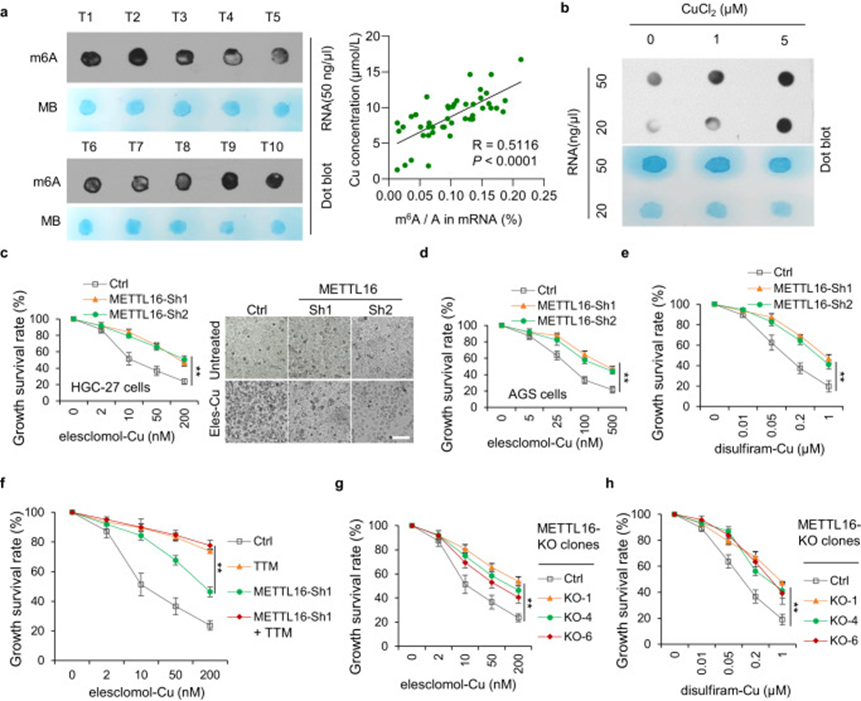
图2 METTL16是铜死亡的重要介导因子
METTL16是一种m6A甲基转移酶,可以修饰多种前序和非编码RNA,促进翻译和癌变,并且是癌症治疗的一个重要潜在靶点。通过TCGA和GTEx数据库的分析发现,在胃癌组织中METTL16的mRNA水平相对较高,而在肿瘤周围组织中较低。免疫组化(IHC)评分结果也显示,胃癌组织中METTL16蛋白水平显著高于正常胃组织。此外,METTL16蛋白水平与TNM分期、病理模式、神经侵袭或血管侵袭无关。有趣的是,高METTL16蛋白水平的胃癌患者的总生存期(OS)和无复发生存期(DFS)显著增加,这与之前的研究结果一致,暗示高METTL16蛋白水平对肿瘤具有潜在的毒性作用。
3. METTL16通过靶向FDX1促进了铜死亡
METTL16负责在许多转录本中进行m6A沉积。因此,作者对稳定的对照-Sh和METTL16-Sh细胞系进行了甲基化RNA免疫共沉淀(MeRIP)测序(图3a)。MeRIP-seq的metagene图显示,在METTL16-Sh细胞中m6A含量减少。使用HOMER软件预测了每个样本中潜在的m6A修饰,并发现m6A修饰主要映射到经典的GGAC基序(图3b)。作者进一步分析了根据MeRIP-seq结果mRNA的总m6A分布模式,发现m6A峰主要富集在CDS和3'UTR区域。KEGG通路分析表明,被METTL16甲基化的基因主要与癌症通路相关。GO富集分析显示,被METTL16修饰的基因富集了与线粒体通路相关的前15个通路(图3c)。此外,作者鉴定了10个与铜死亡相关通路的典型因子,这些因子与低m6A甲基化水平以及METTL16-Sh组中下调的mRNA水平相关(图3d)。qPCR分析显示,METTL16-sh细胞中FDX1、MTF1、PDHB、ATP7A和DLD的mRNA水平下调,其中FDX1在METTL16-sh细胞中表现出最显著的mRNA水平下调(图3e)。此外,在METTL16-Sh或-KO细胞中检测到FDX1蛋白水平的显著下调(图3f,g)。考虑到FDX1是铜离子载体诱导的细胞死亡的关键调节因子,作者推测METTL16诱导的铜死亡依赖于FDX1。
接下来,作者评估了METTL16是否通过影响FDX1的表达来调控铜死亡。鉴于蛋白质硫辛酸化是铜死亡的关键元素,作者评估了METTL16敲除对蛋白质硫辛酸化的影响,使用硫辛酸特异性抗体作为DLAT硫辛酸化的测量标准。结果显示,METTL16敲除导致DLAT硫辛酸化减少,通过免疫印迹进行测量(图3h)。此外,在elesclomol/disulfiram-Cu处理后,FDX1过表达逆转了METTL16敲除和敲除细胞系中铜死亡的拮抗作用(图3i, j)。METTL16过表达诱导的铜死亡通过FDX1敲除得到抑制(图3k)。总体而言,这些数据表明,METTL16通过靶向FDX1促进了铜死亡的发生。
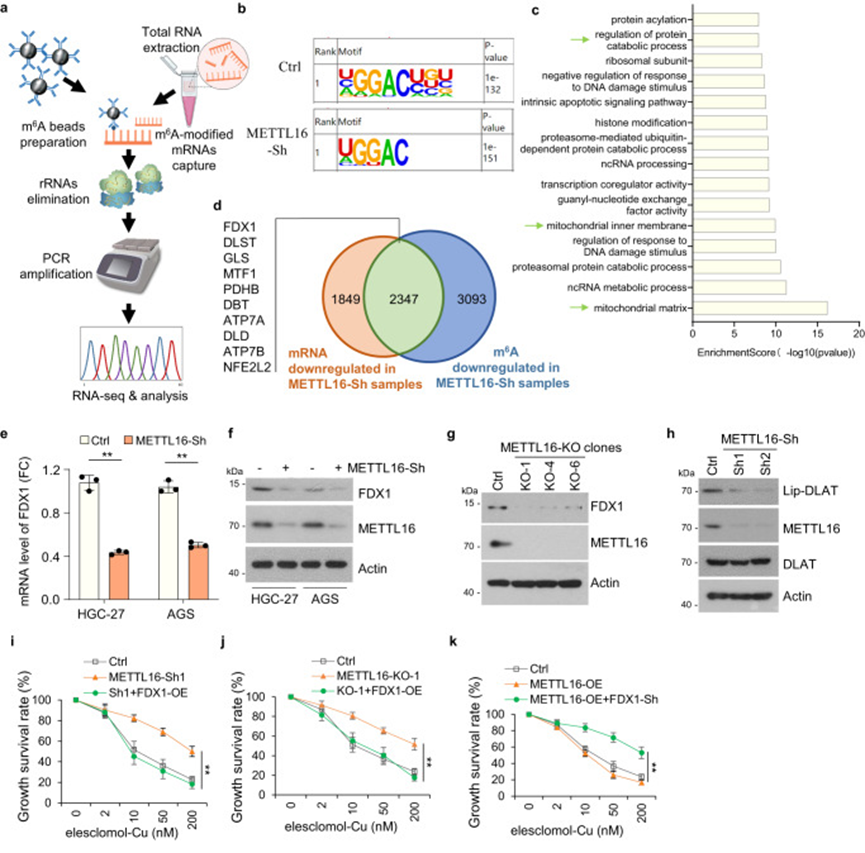
图3 METTL16通过靶向FDX1促进了铜死亡的发生
4. METTL16通过m6A修饰在FDX1 mRNA上促进FDX1蛋白的积累
为进一步探索METTL16如何调控m6A修饰并影响FDX1 mRNA水平,进行了基因特异性MeRIP-qPCR实验。作者发现,在METTL16敲除和敲除细胞中,FDX1 mRNA上的m6A水平显著降低,而在转染METTL16过表达质粒的细胞中显著增加(图4a, b)。当用放线菌D(ActD)停止转录时,FDX1 mRNA在METTL16敲除细胞中的降解速率比对照细胞更快(图4c)。已有报道指出,METTL16位于细胞质促进mRNA翻译,并位于细胞核将m6A沉积到特定RNA靶标29。内源METTL16位于GC肿瘤细胞核中。这些结果表明,METTL16介导的甲基化导致FDX1 mRNA稳定。对FDX1富集m6A峰的IGV分析显示,与对照细胞(红色峰)相比,METTL16-siRNA细胞(绿色峰)中FDX1的m6A水平降低,表明METTL16可能促进FDX1的CDS或3'UTR区域的m6A修饰(图4d)。因此,选取部分CDS区域(选择区域:110456920-110457047和110462353-110462468)以及3'UTR区域(选择区域:110462469-110464884)的FDX1与pGL3荧光素酶报告基因构建FDX1-WT荧光素酶报告基因。共转染FDX1荧光素酶报告基因和不同剂量的METTL16-OE质粒后,发现METTL16显著促进了FDX1-荧光素酶活性(图4e)。
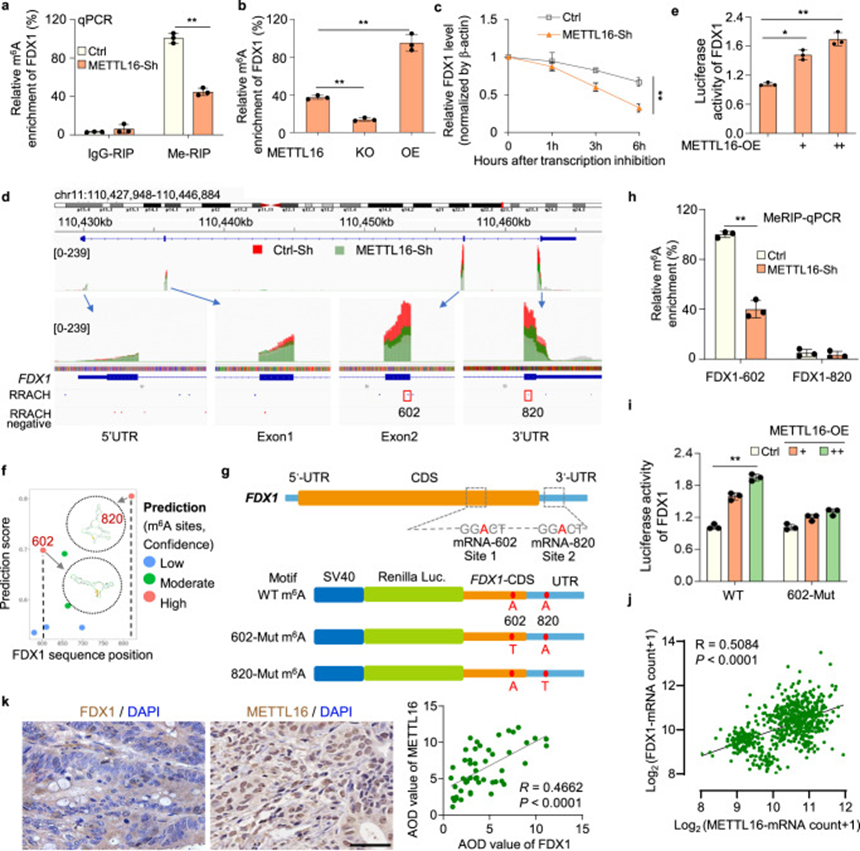
图4 METTL16通过在FDX1 mRNA上进行m6A修饰促进FDX1蛋白的积累。
为进一步探索,作者使用基于序列的m6A修饰位点预测器SRAMP,在FDX1 mRNA上预测了可能的m6A修饰位点。结合预测结果和图4e中的数据,作者确定了两个潜在的m6A修饰位点,具有非常高的置信度:FDX1转录本的CDS区域中的602 A位点和UTR区域中的820 A位点(图4f,g)。随后进行了MeRIP-qPCR分析,使用特定引物证明FDX1转录本上的602位点是METTL16介导的甲基化的直接底物(图4h)。此外,作者设计并构建了FDX1-602-Mut和FDX1-820-Mut荧光素报告基因,通过将m6A基序中的特定腺嘌呤(A)替换为胸腺嘧啶(T),基于FDX1-WT荧光素报告基因(图4g)。双荧光素检测结果表明,METTL16无法促进携带602位点突变的FDX1-CDS/UTR荧光素报告基因的荧光素活性(图4i)。在GEPIA数据库中对METTL16和FDX1的表达进行分析显示,它们在GC中呈正相关(图4j)。另外,免疫组化染色表明,在包含54对GC和相邻正常组织的组织微阵列中,METTL16的表达与FDX1的表达呈正相关(图4k)。这些结果证实了METTL16介导的对FDX1 mRNA的m6A修饰对FDX1 mRNA的稳定性至关重要。
5. METTL16在铜胁迫下发生K229位点的乳酸化
接下来,作者评估了METTL16在GC中是否对铜胁迫作出反应。在铜处理过程中,METTL16的mRNA和蛋白水平保持不变,但FDX1的蛋白水平显著增加(图5a,b)。为验证在铜胁迫下FDX1表达的上调是否依赖于METTL16,作者测量了METTL16敲低或敲除细胞中在有无铜的情况下FDX1蛋白的表达。结果表明,METTL16缺乏阻断了铜诱导的FDX1上调(图5c,d)。由于在铜胁迫下METTL16的mRNA和蛋白水平保持不变,作者随后使用质谱分析测量了METTL16的PTM变化。分析显示METTL16蛋白序列上有十一个乳酸化位点和两个乙酰化位点(图5e)。在乳酸处理后,这六个乳酸化(红色)位点的PTM水平显著增加,暗示这六个位点在肿瘤细胞中可能受到调节。作者随后确认了乳酸化或乙酰化对铜胁迫的响应。结果显示,铜处理显著增加了METTL16的乳酸化水平而非乙酰化水平(图5f)。为了确定METTL16在与铜相关的代谢中的重要乳酸化位点,作者生成了这六个乳酸化位点和K410(作为代表性位点,在乳酸胁迫下乳酸化水平未发生变化)的乳酸化缺陷突变体(K到R)。作者发现只有METTL16-K229R突变体在铜胁迫下显示减少的乳酸化(图5g)。随后,通过LC-MS/MS分析展示了METTL16-K229及其他乳酸化位点的b-y离子匹配图(图5e,图5h)。为进一步确认在铜胁迫下METTL16-K229的乳酸化,作者生成了METTL16 lacty-K229抗体,并使用lacty-K229肽和表达METTL16-WT、-K229R或-K229E的细胞验证了其效力和特异性(图5i,j)。铜和乳酸处理均诱导了METTL16在K229处的强烈乳酸化(图5k,l),表明K229是铜相关代谢中的一个重要乳酸化位点。这些结果表明,在铜胁迫下METTL16在K229位点发生乳酸化。
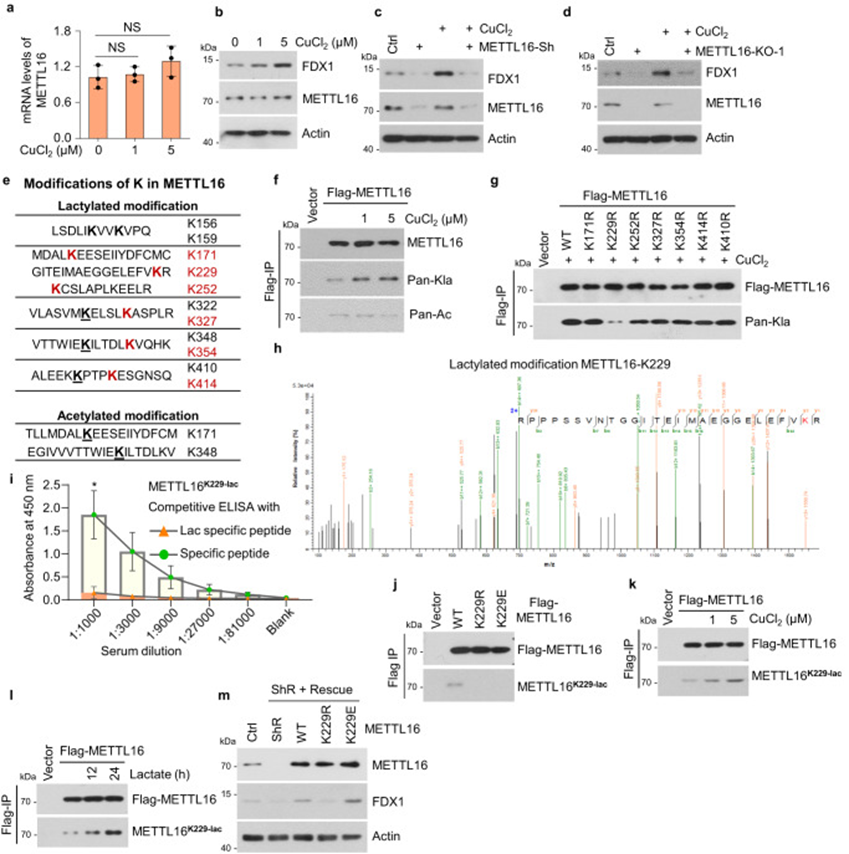
图5 在铜胁迫下,METTL16在K229位点发生乳酸化。
然后,作者分析了K229乳酸化对METTL16的潜在影响。首先,METTL16-K229乳酸化不影响METTL16的核定位。METTL16与MAT2A RNA发夹复合物的晶体结构(PDB 6DU5)显示,K229与E226形成了盐桥,而自我抑制环路中的K163(粉色)插入了METTL16的SAM结合位点。作者的结构建模表明,R230可以通过与环路的K163或Q162形成氢键来稳定自我抑制状态。K229的乳酸化将废除其与E226的盐桥,这可能导致E226侧链的构象变化,并允许与R230形成盐桥。这将削弱自我抑制状态的稳定性,导致METTL16的酶活性升高。接着,作者选择了METTL16的下游靶标,如BCAT1、BCAT2和GPX451来验证METTL16-K229乳酸化对其甲基转移酶活性的影响。结果显示,METTL16-K229E促进了甲基转移酶活性。在稳定的METTL16-恢复细胞系中进一步确保FDX1蛋白水平,其中内源性METTL16被野生型(WT)、乳酸化缺陷型(K229R)或乳酸化模拟型(K229E)所替代。结果显示,在METTL16敲除细胞中FDX1蛋白水平下降,在METTL16-WT或-K229E恢复细胞中恢复,但在METTL16-K229R恢复细胞中没有恢复(图5m)。综上所述,METTL16-K229乳酸化是由铜胁迫诱导的,并促进了METTL16的甲基转移酶活性。
6. SIRT2去乳酸化METTL16-K229并抑制METTL16的活性
以上数据证实了铜激活了METTL16-K229的乳酸化,但肿瘤细胞中涉及的特定去乳酸化酶和乳酸化酶仍不清楚。从理论上讲,乳酸含量越高,可用的乳酸酸就越多,增加蛋白质的乳酸化。因此,作者使用乳酸含量检测试剂盒检测了铜处理的GC细胞内乳酸含量,并发现铜处理后没有显著差异。然后,作者调查铜是否介导了对METTL16的乳酸转移酶或去乳酸酶的调节。进行LC-MS/MS分析以确定METTL16的结合蛋白。作为METTL16的结合蛋白,SIRT2是经典的去乙酰化酶,被分类为主要的去乳酸酶和乳酸转移酶组(图6a)。EIF3b也被确定为METTL16的结合蛋白,与先前的研究结果一致。为了调查和验证METTL16与SIRT2之间的相互作用,作者在HGC-27细胞中检测了METTL16-SIRT2的结合(图6b)。直接METTL16-SIRT2的结合通过His-pull-down实验得到验证。METTL16-K229乳酸化受SIRT2过表达明显抑制,受SIRT2沉默或SIRT2抑制剂AGK2处理促进。此外,铜诱导的METTL16-K229的乳酸化被SIRT2过表达抑制,而被SIRT2沉默或AGK2处理增加。总的来说,这些结果表明,SIRT2与METTL16在K229位点相互作用并对其进行去乳酸化。
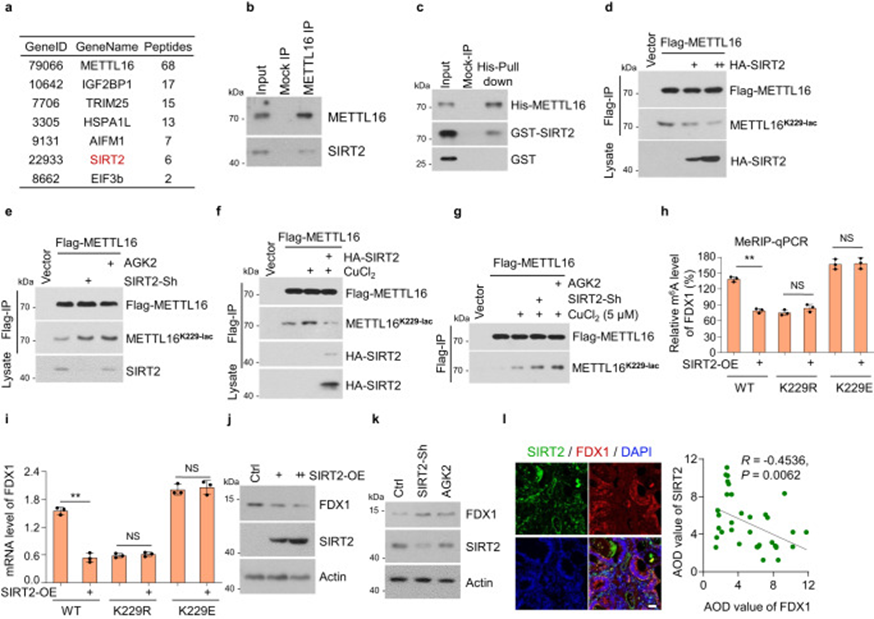
图6 SIRT2去乙酰化METTL16-K229并抑制METTL16的活性。
作者进一步探讨了SIRT2对稳定的METTL16恢复表达细胞系(METTL16-WT、-K229R或-K229E)的调控作用。SIRT2过表达抑制了METTL16-WT细胞中FDX1的m6A水平,但不影响METTL16-K229R或-K229E细胞,表明SIRT2通过METTL16-K229乳酰化抑制了METTL16的活性(图6h,i)。此外,SIRT2过表达抑制了FDX1蛋白水平(图6j),而SIRT2沉默和AGK2处理促进了FDX1蛋白水平(图6k)。免疫荧光染色(IF)的结果表明,在胃癌组织微阵列中,SIRT2蛋白水平与FDX1蛋白水平呈负相关(图6l)。另外,作为去乙酰化酶的SIRT2也可能调节了METTL16的去乙酰化。作者发现,在SIRT2过表达后,总的METTL16乙酰化有所减少。为了检验SIRT2介导的METTL16去乙酰化是否影响METTL16的甲基转移酶活性,作者产生了具有METTL16-Sh恢复的METTL16-WT、-K171R、-K171Q、-K348R和-K348Q的细胞,并进行了m6A水平检测。结果显示,去乙酰化位点对METTL16的甲基转移酶活性没有影响。此外,在METTL16-WT、-K171R、-K171Q、-K348R和-K348Q过表达后,FDX1 mRNA水平的增加没有显著差异。这些数据表明,SIRT2介导的METTL16去乙酰化并不影响FDX1 mRNA水平。在SIRT2敲除或AGK2处理下,METTL16的表达与否没有显著差异。综上所述,这些结果表明,SIRT2通过在K229位点进行去乳酰化来负向调节METTL16的活性。
为了进一步探讨铜离子对SIRT2对METTL16的调控作用,作者检测了铜离子是否影响SIRT2和METTL16的相互作用。令人意外的是,结果显示铜处理略微减少了SIRT2和METTL16的结合。考虑到在铜离子处理下METTL16的乳酰化增加,作者有理由推测铜离子处理可能增强了METTL16和乳酰基转移酶的结合。
7. SIRT2-METTL16-FDX1-铜离子诱导的细胞凋亡轴支持一种有前景的治疗策略
FDX1是铜离子诱导的细胞凋亡的重要介体,这是一种治疗癌症的潜在方法。作者发现,在SIRT2抑制或铜诱导的METTL16-K229乳酰化后,FDX1蛋白水平增加。接下来,作者检查了METTL16-K229乳酰化对铜离子诱导的细胞凋亡的影响。结果显示,在铜应激条件下,METTL16敲除导致的DLAT硫辛酰化水平在METTL16-WT/-K229E细胞中得到了恢复,但在METTL16-K229R细胞中未能实现(图7a)。此外,在铜离子和elesclomol处理条件下,METTL16-WT或-K229E细胞诱导了细胞凋亡,但METTL16-K229R细胞未见此效应(图7b)。此外,SIRT2过表达逆转了METTL16-WT在铜应激条件下所诱导的DLAT硫辛酰化增加,但不包括METT16-K229E,表明SIRT2通过去乳酰化METTL16-K229抑制了细胞凋亡(图7c)。一致地,AGK2处理促进了在铜应激条件下METTL16-WT细胞中的DLAT硫辛酰化,但在METTL16-K229R细胞中未见此效应(图7d)。此外,在elesclomol和铜离子存在的情况下,AGK2处理促进了细胞凋亡(图7e)。
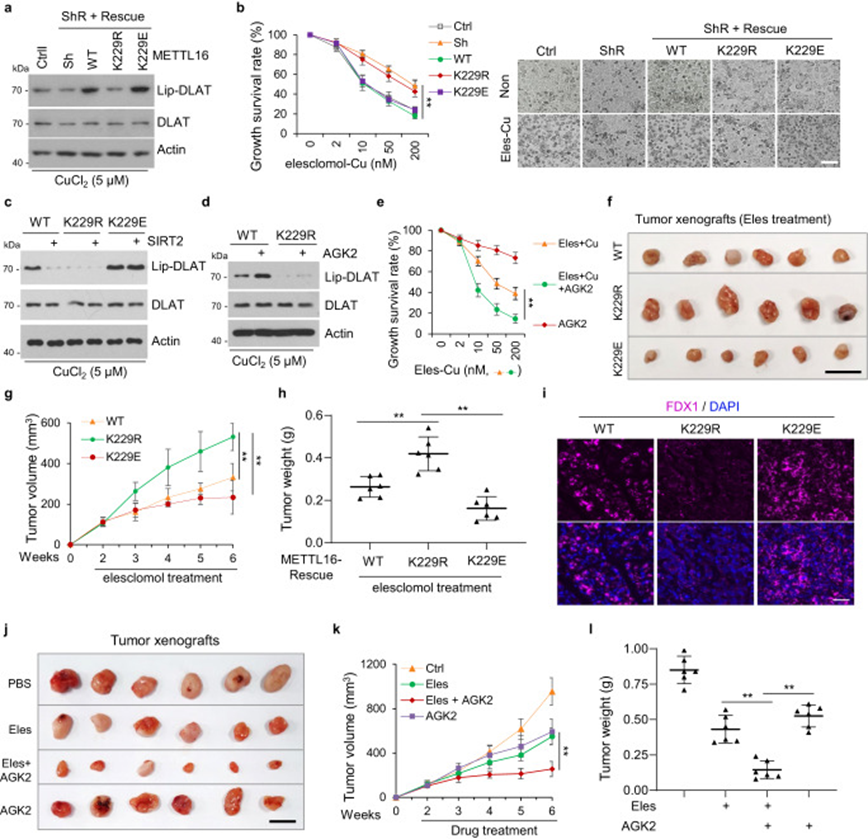
图7 SIRT2-METTL16-FDX1-铜凋亡轴支持一种有前景的治疗策略。
作者进一步评估了METTL16的乳酸化是否可以增强接受elesclomol治疗的小鼠异种移植肿瘤的治疗反应。METTL16-WT、-K229R和-K229E细胞被皮下注射到裸鼠的左右后腿上方。观察到在接受elesclomol治疗的小鼠中,METTL16-WT和-K229E组的肿瘤生长明显受到抑制,但在METTL16-K229R组中未观察到这种情况,表现为肿瘤体积和重量的减少(图7f–h)。
肿瘤切片中的FDX1染色显示,在肿瘤异种移植中,METTL16-WT和-K229E组中的FDX1染色较METTL16-K229R组更高(图7i)。这些结果表明,在K229位点的METTL16的乳酸化状态在决定铜凋亡中发挥了至关重要的作用。此外,在肿瘤异种移植中采用了elesclomol和AGK2的联合治疗。肿瘤切片的分析显示,在体内AGK2增强了elesclomol的治疗效果,表现为肿瘤体积和重量的减少(图7j–l)。这些结果表明elesclomol和AGK2的联合治疗在胃癌中是一种有前景的治疗方法,特别适用于恶性肿瘤——粘液腺癌(铜含量较高)。
实验方法
细胞培养;小鼠异种移植肿瘤实验;RNA提取和实时荧光定量聚合酶反应(qRT-PCR);荧光素酶报告基因检测;蛋白质印迹;His pull-down 检测;点突变构建;shRNA/siRNA设计和构建;铜微孔板测定;免疫沉淀 (IP);免疫荧光染色;MeRIP-qPCR酶;甲基化 RNA 免疫沉淀测序 (MeRIP-seq);RNA 免疫沉淀 (RIP) 检测;RNA稳定性测定;CRISPR/Cas9敲除细胞系;体外乳酰化试验;生物信息学分析
参考文献
Sun L, Zhang Y, Yang B, Sun S, Zhang P, Luo Z, Feng T, Cui Z, Zhu T, Li Y, Qiu Z, Fan G, Huang C. Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer. Nat Commun. 2023 Oct 20;14(1):6523.