






METTL17通过调节结直肠癌中的线粒体翻译来协调铁死亡和肿瘤发生
铁死亡是一种铁依赖的脂质过氧化诱导的细胞调节性死亡,在癌症治疗中显示出优良的前景。尽管线粒体在铁死亡的调控中发挥重要作用,但其潜在机制仍不清楚。本研究揭示了线粒体蛋白METTL17通过表观遗传调控调控结直肠癌(CRC)细胞的线粒体功能。生物信息学分析证实METTL17的表达与癌细胞的铁死亡抵抗呈正相关,并且在CRC中上调。METTL17的缺失使CRC细胞对铁死亡敏感,损害细胞增殖,迁移,侵袭,异种移植肿瘤生长,以及AOM/DSS诱导的CRC肿瘤发生。此外,在铁死亡应激过程中,METTL17的抑制会破坏线粒体功能和能量代谢,并增强细胞内和线粒体脂质过氧化和ROS水平。在机制上,抑制METTL17可显著降低线粒体RNA甲基化,包括m4C、m5C、m3C、m7G和m6A,导致线粒体蛋白编码基因的翻译受损。此外,与METTL17相关的相互作用蛋白对线粒体基因表达至关重要,敲低它们可使CRC细胞对铁死亡敏感并抑制细胞增殖。值得注意的是,联合靶向METTL17和铁死亡的治疗方法有效地抑制了体内结直肠癌异种移植的生长。本研究揭示了METTL17介导的线粒体中细胞存活和铁死亡的防御机制,突显了METTL17是治疗CRC的潜在靶点。本文于2024年2月发表在《Redox Biology》,IF:11.4。
技术路线:
主要实验结果:
1.METTL17的表达与CRC细胞对铁死亡的抵抗呈正相关,敲低METTL17可使CRC细胞对铁死亡增敏
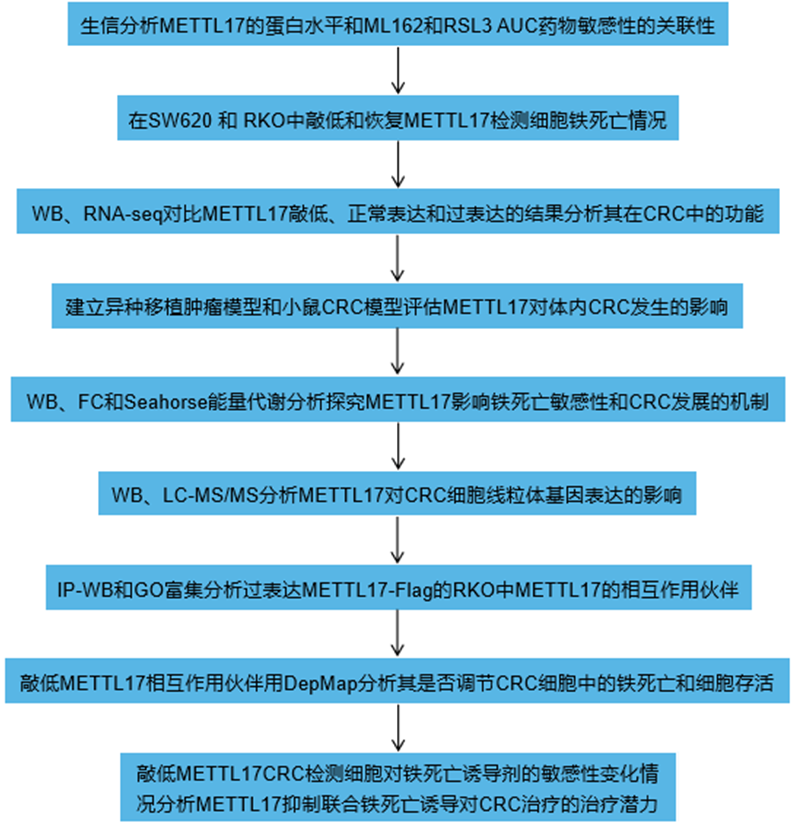
癌症治疗反应门户(CTRP)提供了一个包含545种化合物处理的907个癌细胞系的基因表达和耐药信息的综合数据集。值得注意的是,对CTRP数据的分析表明,METTL17 mRNA表达与对GPX4抑制剂(包括ML162和RSL3这两种铁死亡的经典诱导剂)的耐药性呈正相关(图1A)。进一步对另外两个数据库,癌症依赖图(DepMap)和癌症细胞系百科全书(CCLE)的综合分析表明,在各种癌细胞株中METTL17的蛋白水平与ML162和RSL3 AUC药物敏感性呈正相关(图1B和C)。
为了确定METTL17是否调控CRC的铁死亡,利用慢病毒介导的shRNA敲低METTL17构建稳定细胞株。与在CTRP、DepMap和CCLE数据集中观察到的相关性一致,在SW620和RKO这两个高水平表达METTL17的结直肠癌(CRC)细胞中敲低METTL17(图2D),增加了对ML162或RSL3诱导的铁死亡的敏感性,而细胞活力被铁螯合剂去铁胺(DFO)挽救(图1D和E)。此外,碘化丙胺(PI)染色显示,ML162或RSL3处理的METTL17缺陷细胞表现出更多的细胞死亡,而DFO能显著逆转(图1F-H),表明敲低METTL17使CRC细胞对GPX4抑制敏感。
接下来,作者在METTL17敲低的SW620细胞中外源性表达METTL17,发现恢复METTL17表达部分挽救了ML162诱导的铁死亡。然而,在Scramble细胞中过表达METTL17轻微促进了ML162诱导的铁死亡。此外,METTL17敲低加速了Erastin(胱氨酸/谷氨酸反向转运体系统Xc-的抑制剂)诱导的铁死亡。在METTL17敲低的RKO细胞中,恢复METTL17的表达部分挽救了Erastin诱导的铁死亡。总之,这些生物信息学和实验结果证实了METTL17在CRC铁死亡调节中的重要作用。
图一:METTL17的表达与CRC细胞的铁死亡抵抗呈正相关,敲低METTL17可增加CRC细胞对铁死亡的敏感性。
2.2.METTL17在人结直肠癌中高表达,敲低METTL17可抑制结直肠癌细胞的增殖、迁移、侵袭和体外致癌特征
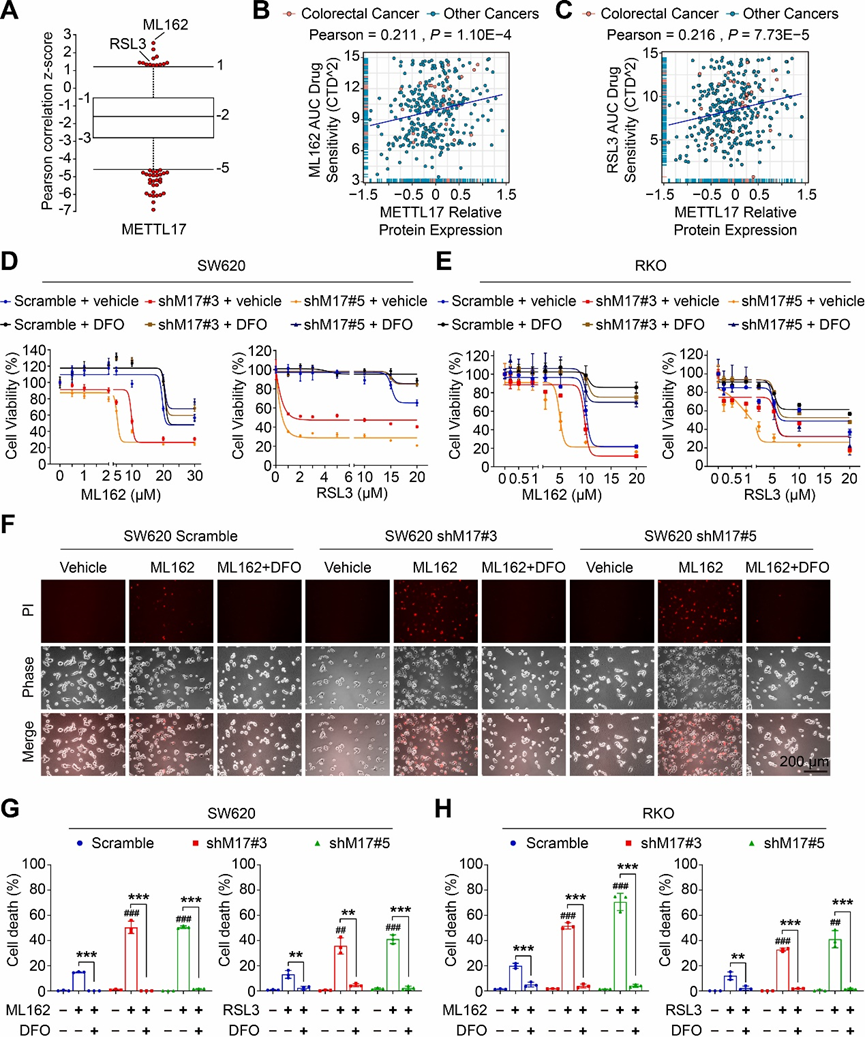
癌症基因组图谱(TCGA)分析发现,与正常结直肠组织相比,CRC组织中的METTL17 mRNA上调(图2A)。相应的,METTL17蛋白在CRC组织中的表达高于其相邻的正常组织(图2B和C)。随后研究人员评估了METTL17在CRC中的功能。首先,基于DepMap和CCLE数据库,作者观察到METTL17 mRNA表达水平在所有CRC细胞系中的分布不同,也表明了METTL17在CRC中的表达与其基因效应之间的相关性。其次,与正常结肠上皮细胞系相比,METTL17蛋白水平在CRC细胞系中显著升高(图2D)。在CRC细胞系中,RKO、SW620和DLD1具有较高的METTL17表达,因此选择它们进行后续的表型实验。
为了确定METTL17在CRC细胞进展中的作用,研究人员使用两种不同的针对METTL17的短发夹RNA敲低了三个人CRC细胞系SW620、RKO和DLD1中的METTL17表达,并通过Western blot证实了敲低效率(图2E)。结晶紫实验显示METTL17敲低CRC细胞中细胞增殖的显著抑制(图2F)。此外,细胞迁移和侵袭实验表明,在RKO、SW620和DLD1细胞中,METTL17的下调显著抑制了它们的跨膜迁移和通过基质包膜侵袭的能力(图2G),表明METTL17敲低阻碍了CRC细胞的迁移和侵袭。细胞集落形成分析进一步表明,敲低METTL17能显著抑制CRC细胞生长(图2G)。此外,细胞周期分析表明,敲低METTL17导致G0/G1期延长和S期减少,表明在METTL17缺陷的CRC细胞中,G0/G1期的细胞周期停滞增加(图2H)。
为了阐明METTL17调控CRC细胞存活的机制,研究人员对METTL17敲低和野生型CRC细胞进行了RNA-seq分析,然后进行了基因集富集分析(GSEA)。与观察到的细胞周期停滞在G0/G1期的METTL17缺陷细胞相一致,GSEA显示,与对照细胞相比,METTL17缺陷的CRC细胞在细胞周期阶段转换(图2I)和DNA复制(图2J)方面存在缺陷。此外,根据GSEA数据研究人员发现许多致癌特征基因集因METTL17缺陷而下调(图2K)。受损的致癌基因包括ERBB2通路、LEF1通路、KRAS通路和MEK通路,其中,致癌基因的表达因METTL17基因敲除而下调。这些发现共同表明,敲低METTL17能显著抑制CRC细胞的致癌活性。
此外,研究人员还发现恢复METTL17的表达部分逆转了在METTL17敲低的SW620细胞中观察到的受抑制的细胞增殖(图2L)。综上所述,这些结果强调了METTL17在人类CRC中过表达,并且敲低其表达可以抑制CRC细胞的增殖,迁移,侵袭,集落形成和细胞周期转变,至少部分是通过破坏致癌通路来实现的。
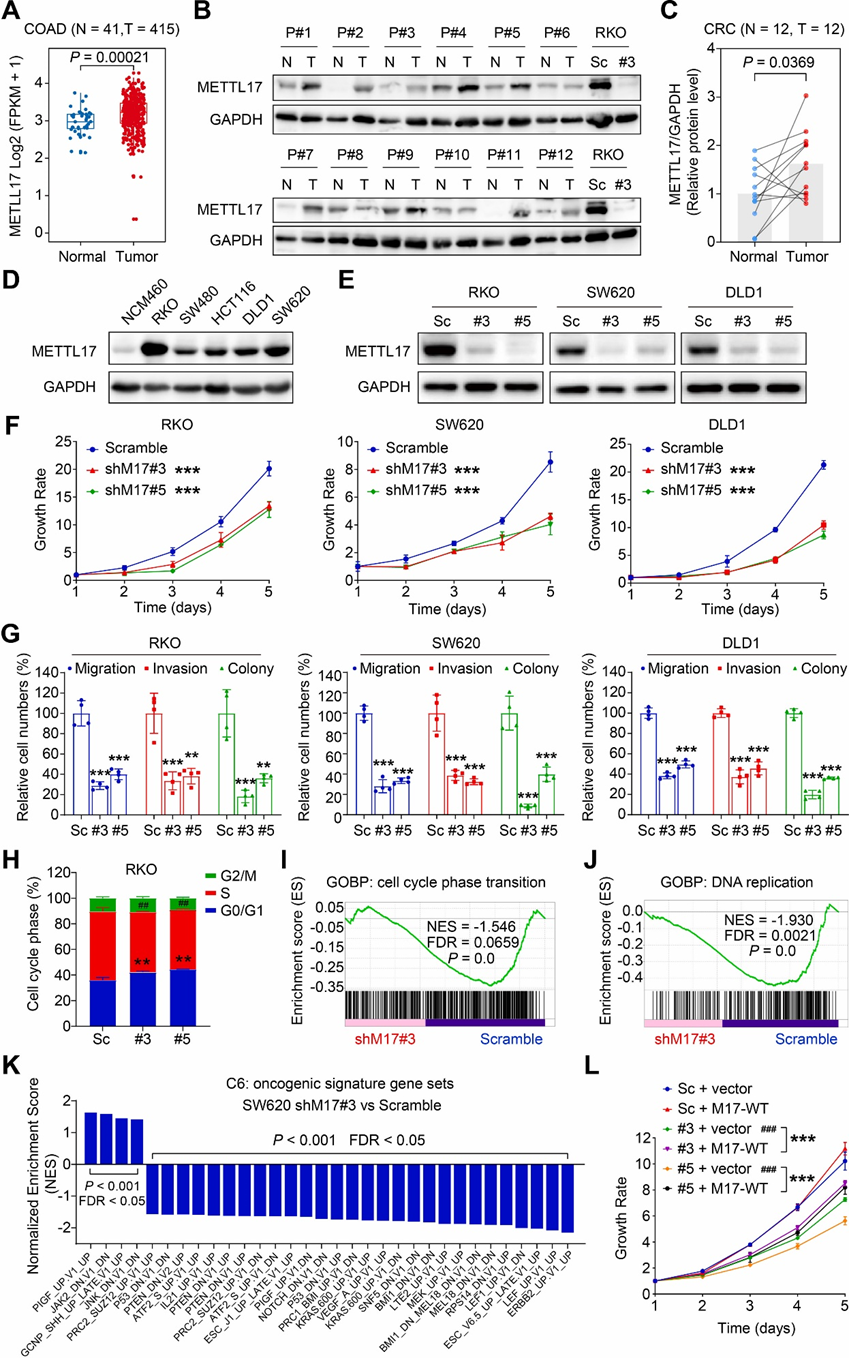
图2:METTL17在CRC中高表达,体外敲低METTL17可抑制CRC细胞的增殖、迁移、侵袭和致癌特征。
3.METTL17缺陷抑制异种移植瘤的生长和AOM/DSS诱导的体内结直肠癌的发生
为了评估METTL17对体内CRC发生的影响,研究人员进行了异种移植肿瘤模型和AOM/DSS诱导的小鼠结肠炎相关的CRC模型。首先,将METTL17敲低的SW620细胞和对照SW620细胞分别注射到裸鼠皮下。结果显示,METTL17敲低的肿瘤比对照肿瘤生长得慢得多,重量也小得多(图3A、B、C)。Western blot和免疫染色分析表明,METTL17敲低的肿瘤显示出β-Catenin的表达减少,而PCNA的蛋白水平和Ki-67阳性细胞的数量保持不变(图3D、E、F)。这些结果表明,在异种移植肿瘤模型中,敲低METTL17通过非增殖依赖性途径抑制肿瘤生长。
其次,研究人员通过AOM/DSS治疗诱导了结肠炎相关的CRC。鉴于在METTL17敲除小鼠中观察到的死亡率,实验使用了METTL17杂合子(METTL17+/-),这些杂合子在结肠上皮中显示出METTL17表达减少,而在结肠发育中没有观察到缺陷。有趣的是,研究人员发现METTL17+/-保护了小鼠,使其免受AOM/DSS诱导的CRC肿瘤发生,这可以通过肿瘤大小和肿瘤数量的减少得到证明(图3G和H)。在METTL17+/-CRC样本中,PCNA和β-Catenin的蛋白表达水平下调(图3I和J)。接下来,研究人员通过对WT和METTL17+/-小鼠的结直肠肿瘤进行Ki-67和β-Catenin免疫染色,评估了细胞增殖率和恶性级别。结果表明,METTL17缺陷抑制了肿瘤进展,因为METTL17+/-小鼠表现出具有分化良好的腺管结构和β-Catenin弥漫性胞质分布的良性腺瘤,而WT小鼠表现出具有低分化癌和β-Catenin核定位的恶性肿瘤(图3K)。这些结果表明,METTL17缺陷抑制异种移植肿瘤的生长和AOM/DSS诱导的体内结直肠癌进展。
图3.缺乏METTL17可减少异种移植肿瘤的生长和AOM/DSS诱导的体内CRC肿瘤发生。
4.敲低METTL17会破坏线粒体稳态,促进铁死亡应激下的脂质过氧化和ROS生成
为了阐明METTL17影响铁死亡敏感性和CRC发展的机制,作者研究了METTL17在癌细胞中的分布和功能。首先将RKO细胞分为线粒体和细胞质两部分,通过Western blot检测两部分的内源性METTL17蛋白水平和细胞定位。研究结果表明METTL17主要在线粒体中表达(图4A),这与之前的报道一致,即METTL17由核基因组编码,是一种具有N端转运肽的线粒体蛋白。其次,由于现有的METTL17抗体均不符合研究人员的免疫荧光质量标准,因此研究人员生成了一个C末端Flag标记的METTL17构建体,并在CRC细胞中过表达(图4B)。在稳定表达3xHA-EGFP标记线粒体的RKO和SW620细胞系中外源性过表达METTL17-C-Flag后,通过荧光共聚焦显微镜观察到METTL17-C-Flag在线粒体中的大量定位(图4C)。因此,这些结果证实了METTL17的线粒体定位。
为了研究METTL17在线粒体中的功能,研究人员检测了METTL17敲低细胞中能量代谢的变化。最初,研究人员测定了线粒体氧耗率(OCR),并观察到METTL17敲低后OCR显著受到抑制,表现为基础呼吸、ATP生成、最大呼吸和备用容量减少,尤其是在shM17#5细胞中(图4D)。GSEA分析一致表明,在METTL17缺陷细胞中,与氧化磷酸化相关的基因下调(图4E),表明METTL17敲低导致线粒体功能障碍。随后,研究人员评估了细胞外酸化率(ECAR),发现敲除METTL17的细胞糖酵解能力减弱,糖酵解储备减少(图4F)。此外,研究人员还发现敲低METTL17会显著抑制癌细胞的葡萄糖摄取能力(图4G),表明METTL17缺失细胞的葡萄糖供应受损。综上所述,这些发现揭示了METTL17在维持线粒体稳态和糖酵解中的关键作用。
由于ROS和氧化脂质的积累是铁死亡的决定因素,因此线粒体事件在这一受调节的细胞死亡过程中起着至关重要的作用。随后,研究人员使用四种荧光探针分别通过流式细胞术和激光共聚焦显微镜评估活细胞中ROS和氧化脂质的细胞和线粒体水平。ML162处理3h后,SW620细胞脂质过氧化物含量增加,METTL17缺陷显著增加铁死亡胁迫下脂质过氧化物的形成。C11-BODIPY和MitoPerOx的红色荧光转变为绿色荧光(图4H),表明METTL17敲低的癌细胞在铁死亡应激过程中存在过度的细胞内和线粒体脂质过氧化。同时,如DCFH-DA和MitoSOX染色所示,在ML162处理后,METTL17敲低的癌细胞中,细胞内和线粒体ROS水平显著升高(图4I)。
此外,通过透射电子显微镜发现,METTL17敲低细胞中存在显著的线粒体损伤,表现为ML162攻击后线粒体收缩增加和嵴减少(图4J和K)。研究人员接下来通过检测关键铁死亡标志物的蛋白水平,研究了METTL17调节铁死亡的机制。令人惊讶的是,METTL17敲低显著抑制了血红素加氧酶1(HMOX1/HO-1)的表达,并显著上调了ML162处理后NRF2的表达(图4M)。然而,在mettl17敲低的细胞中,铁死亡的关键调节因子,包括xCT、GPX4和ACSL4,在基础和ML162处理条件下保持不变(图4M)。此外,我们发现单独敲低METTL17足以增加NADH水平(图4L)。然而,在铁死亡应激下,METTL17敲低加速了NADH的下降(图4L),表明在铁死亡期间,METTL17缺陷细胞中的抗氧化系统被破坏。此外,单独敲低METTL17并不影响氧化还原稳态相关基因的表达。总之,这些结果表明,METTL17介导的线粒体稳态对维持CRC生长和抵御铁死亡至关重要。
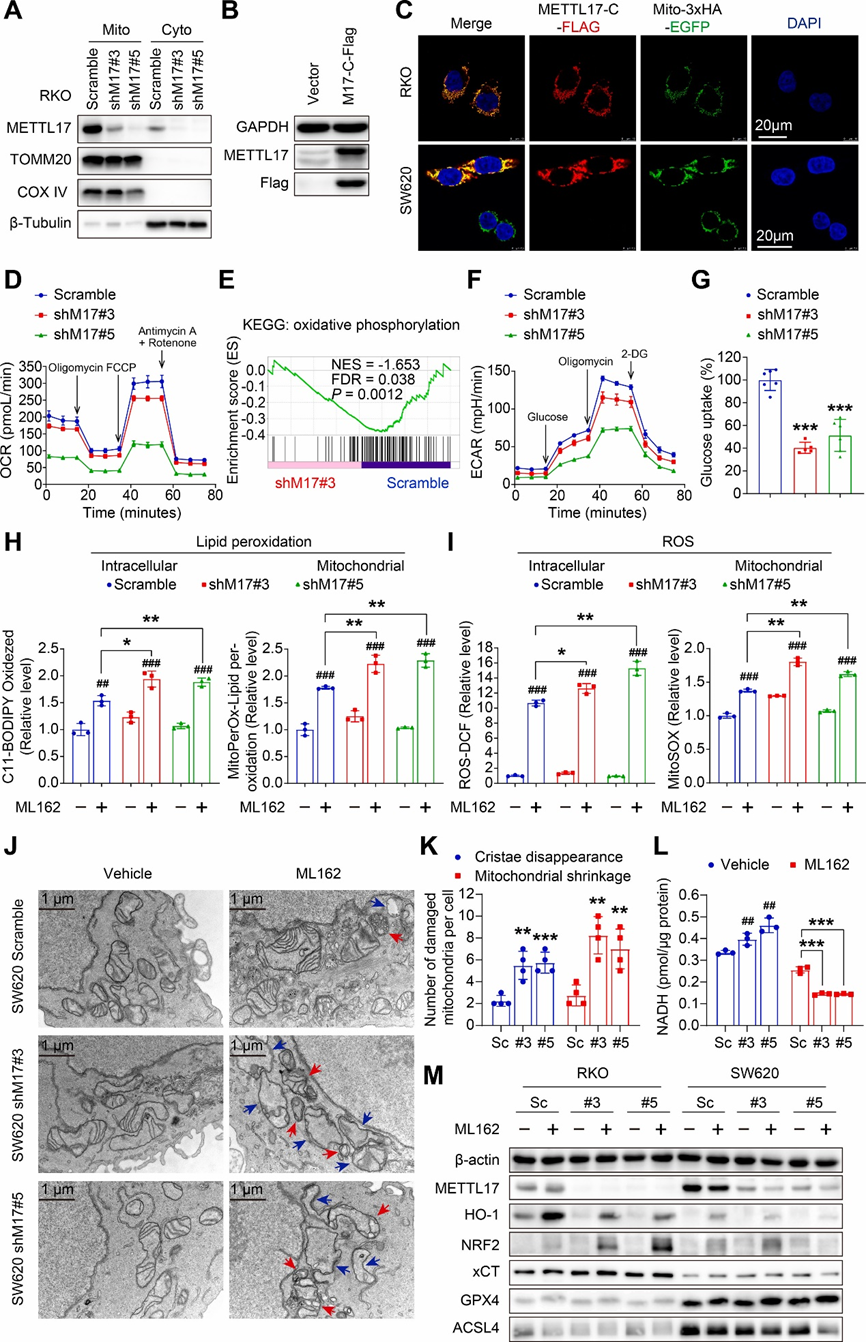
图4.METTL17的缺失会导致线粒体功能严重失调,并促进铁死亡线粒体脂质过氧化。
5.METTL17通过线粒体RNA甲基化的方式控制CRC的线粒体翻译
METTL17被认为是mESCs中线粒体基因表达的调节因子。然而,目前尚不清楚METTL17是否是癌细胞中线粒体基因表达所必需的。首先,研究人员对DepMap数据库中METTL17的前100个共依赖基因进行了全面分析,DepMap数据库通过全基因组汇总的CRISPR-Cas9敲除筛选,对700多个癌症细胞系的基因依赖性进行了评分,并对这些基因进行了基因本体(GO)分析。GO分析显示METTL17共依赖基因编码蛋白显著富集于线粒体内膜和线粒体核糖体。这些蛋白在协调线粒体基因表达和翻译中发挥不可或缺的作用,影响多种过程,如线粒体体生物发生、NADH、泛醌和醌活性(图5A)。为了验证METTL17参与线粒体基因调控,研究人员使用Western blot检测了13个线粒体蛋白编码基因的蛋白水平。结果表明,在CRC细胞中,METTL17敲低导致CRC细胞中线粒体蛋白的整体减少(图5B和C),然而,mRNA水平没有同时发生改变。这表明METTL17对于维持癌细胞中正常的线粒体基因表达是必需的。
虽然之前的研究表明,METTL17与12S mt-rRNA结合并介导mESCs中m4C840和m5C842甲基化,但其在其他甲基转移酶活性,特别是在mt-tRNAs和mt-mRNAs中的潜在作用仍不清楚。为了填补这一知识缺口,研究人员对线粒体RNA进行了LC-MS/MS分析,以探索METTL17敲低的CRC细胞中甲基化模式的变化(图5D)。研究结果显示,与对照细胞相比,shM17#5中m4C水平显著降低至约20%,而与METTL17相关的另一种RNA修饰m5C在METTL17敲低细胞中降低至约75%(图5E)。有趣的是,m3C(先前与mt-tRNA中的METTL8相关的修饰)在METTL17敲低的细胞中下降到大约70%(图5E),这表明METTL17可能是mt-tRNA中m3C的一个尚未确定的甲基化调节因子。此外,敲低METTL17也降低了m7G和m6A的水平(图5E),表明METTL17可能是线粒体RNA甲基化的多功能调节因子。重要的是,敲低METTL17不影响其他线粒体甲基转移酶的表达。综上所述,表明METTL17通过调节CRC细胞中12S mt-rRNA中的m4C和m5C,mt-tRNA中的m3C和m7G,以及mt-mRNA中的m6A的甲基化来调控线粒体基因的表达。
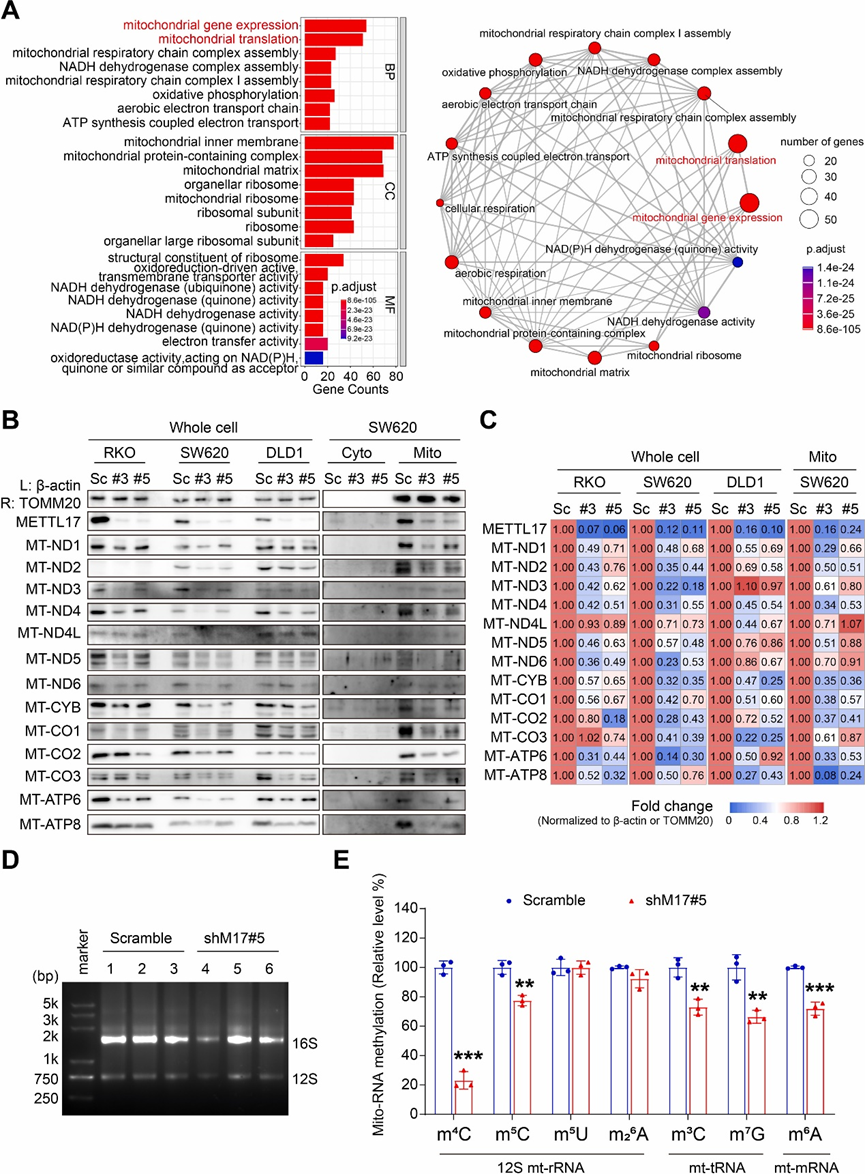
图5:在CRC中,METTL17是线粒体RNA甲基化方式下全局线粒体翻译所必需的。
6.与METTL17相互作用的蛋白调控线粒体基因表达和铁死亡
多种蛋白有利于线粒体基因表达和蛋白翻译。为了揭示METTL17的相互作用伙伴,在慢病毒过表达METTL17-Flag的RKO细胞中,分别用Flag免疫沉淀(IP)富集线粒体、细胞质和全细胞蛋白裂解物(图6A)。通过质谱(MS)对富集的蛋白质进行鉴定,以覆盖度大于50%和唯一肽段大于20为标准进行数据过滤。人类线粒体蛋白质组数据库Human MitoCarta包含1136个同种人线粒体蛋白质,利用该数据库生成的韦恩图显示了26个与METTL17具有高亲和力的线粒体蛋白(图6B)。GO富集分析表明,这些蛋白在线粒体基因表达和线粒体翻译中发挥重要作用。值得注意的是,IP-Western blot分析显示METTL17与MRPS9,MRPS22和MRPS35之间有很强的亲和力,而与MRPL15和MRPL11之间的相互作用较弱(图6C)。这表明METTL17与线粒体核糖体的小亚基(MSSU),而不是线粒体核糖体的大亚基(MLSU)之间存在特定的相互作用。除了MSSU,研究还发现METTL17和LRPPRC之间存在高度相互作用(图6C),LRPPRC是一种线粒体蛋白,对线粒体mRNA的多聚腺苷酸化和翻译协调至关重要。
为了探索METTL17相互作用蛋白是否也调节CRC细胞中的铁死亡和细胞存活,研究人员在RKO细胞中使用慢病毒shRNA敲低LRPPRC、MRPS9、MRPS22和MRPS35。值得注意的是,与METTL17敲低实验相似,敲低这些METTL17相互作用蛋白使癌细胞对ML162诱导的铁死亡敏感,并抑制CRC中的细胞增殖(图6D-G)。DepMap分析显示,METTL17和LRPPRC、MRPS9、MRPS22和MRPS35之间的基因效应具有强相关性,突出了METTL17相关的细胞功能在癌细胞生存和铁死亡防御中的重要性(图6D-G)。CTRP和DepMap分析表明,LRPPRC、MRPS9、MRPS22和MRPS35的表达升高与对ML162和RSL3的耐药性相关。此外,敲低METTL17并不影响这些相互作用蛋白的表达。值得注意的是,这些发现揭示了METTL17相互作用蛋白在控制癌细胞生存方面的高度相关性。
综上所述,这些结果确定了METTL17及其相互作用蛋白共同调节线粒体基因的表达,从而有助于CRC细胞的铁死亡防御和细胞存活。
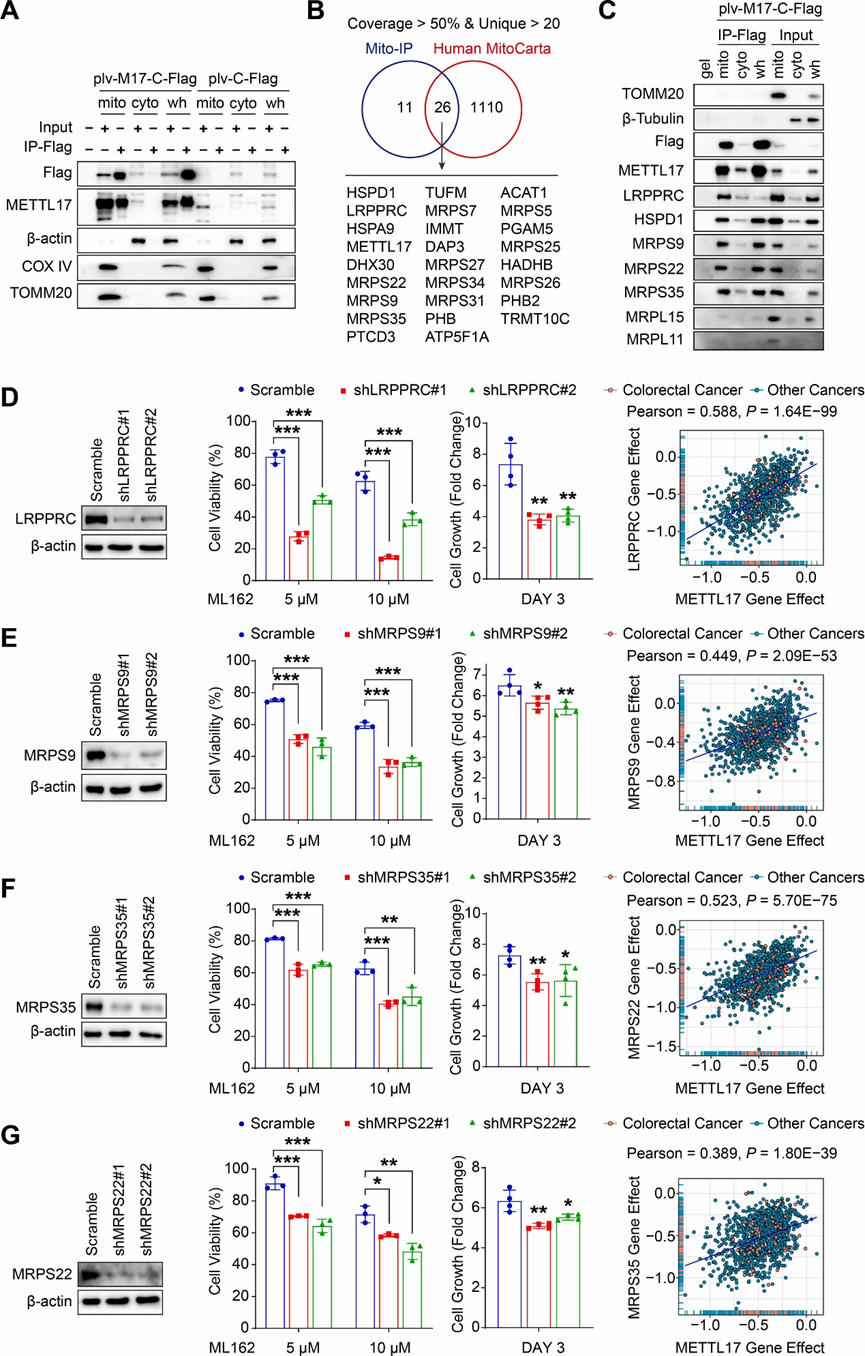
图6:靶向METTL17相互作用蛋白使癌细胞对铁死亡敏感
7.靶向METTL17和铁死亡的联合治疗抑制CRC肿瘤的发生
由于单独敲低METTL17在体内抑制了SW620异种移植瘤的生长,并且在体外使CRC细胞对GPX4抑制敏感,研究人员进一步研究了联合METTL17抑制和铁死亡诱导对CRC治疗的治疗潜力。首先,研究人员研究了敲低METLL17是否增加CRC细胞对铁死亡诱导剂的敏感性。事实上,METTL17敲低使SW620异种移植瘤对ML162治疗增敏,表现出对CRC治疗的协同作用,这一点可以从肿瘤体积、大小和重量的减小中得到证明(图7A和B)。重要的是,与利普司他丁-1(Lip-1,一种通过阻断脂质过氧化而建立的铁死亡抑制剂)联合治疗减弱了ML162治疗下METTL17敲低肿瘤的生长抑制作用(图7A和B)。ML162处理不影响这些肿瘤中的Ki-67染色,但与对照肿瘤相比,在METTL17敲低的肿瘤中,4-HNE染色(脂质过氧化的标志物)显著增加(图7C和D)。相反,在ML162处理的METTL17敲低的肿瘤中,给药Lip-1减弱了脂质过氧化的增强(图7C和D)。这些结果表明,同时抑制METTL17和铁死亡诱导剂可通过增强铁死亡在CRC治疗中提供协同效应。
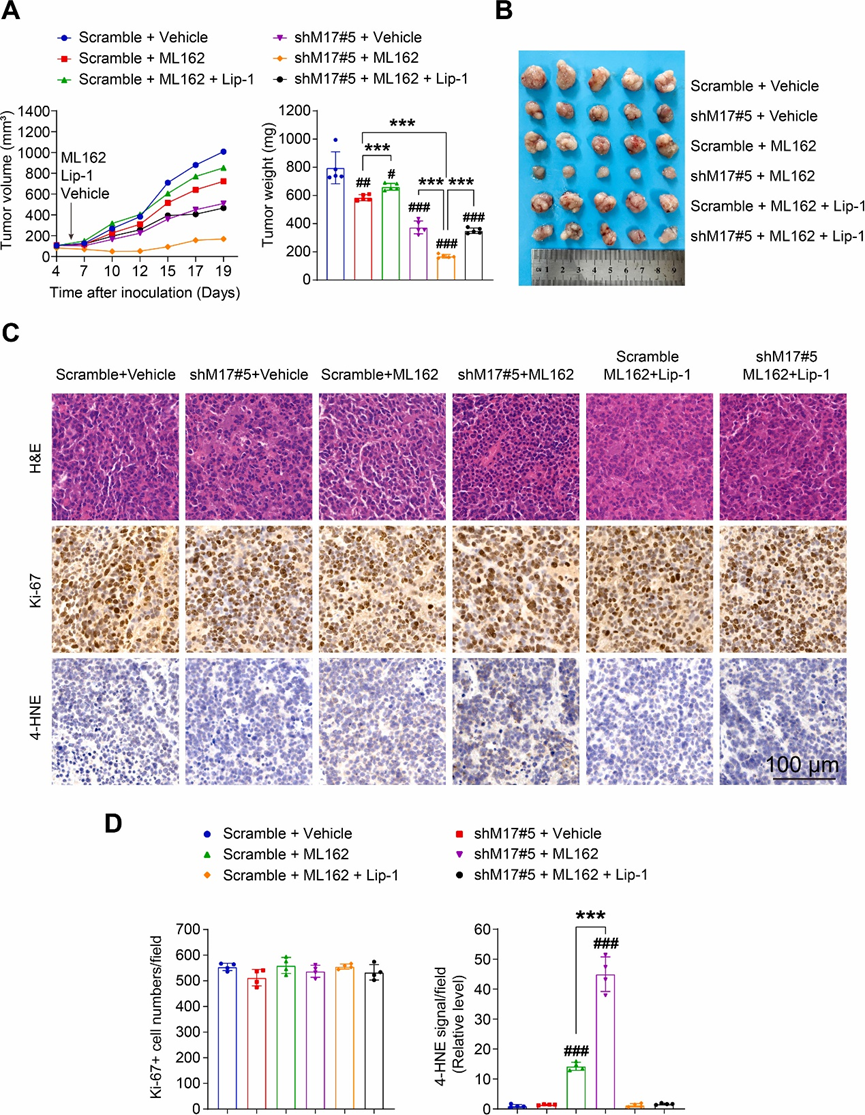
图7:靶向METTL17和铁死亡的联合治疗可抑制CRC肿瘤的发生
实验方法:
抗体和蛋白免疫印迹、跨孔迁移和侵袭试验、建立小鼠模型、异种移植肿瘤生长实验、免疫组化分析、免疫荧光、RNA测序、线粒体RNA甲基化检测、Seahorse能量代谢分析
参考文献:
Li H, Yu K, Hu H, Zhang X, Zeng S, Li J, Dong X, Deng X, Zhang J, Zhang Y. METTL17 coordinates ferroptosis and tumorigenesis by regulating mitochondrial translation in colorectal cancer. Redox Biol. 2024 Feb 13;71:103087. doi: 10.1016/j.redox.2024.103087. Epub ahead of print. PMID: 38377789; PMCID: PMC10884776.