






TRMT10C调控的m1A甲基化与阿尔茨海默病中的线粒体功能失调有关
线粒体ND5 mRNA有一个m1A位点。ND5是呼吸链复合体I的一个亚基。它被认为是氧化和质子传输耦合的关键。在这里,作者证明了这个m1A位点可能参与阿尔茨海默病(AD)的病理生理学。这种神经退行性疾病的病理特征之一是线粒体功能障碍,主要由淀粉样β(Aβ)引起。Aβ主要干扰呼吸链复合体I和IV的功能。然而,复合物I功能障碍的分子机制仍不完全清楚。作者在AD细胞模型和AD患者中发现ND5 mRNA的m1A甲基化增强。这种m1A甲基化的形成是由TRMT10C蛋白水平的增加催化的,导致ND5翻译抑制。因此,本文首次证明,TRMT10C诱导的ND5 mRNA的m1A甲基化导致线粒体功能障碍。该研究结果表明,这一新发现的机制可能与Aβ诱导的线粒体功能障碍有关。该研究于2024年1月发表在《molecular psychiatry》,IF 11。
技术路线:
主要研究结果:
1 AD细胞模型和AD患者TRMT10C mRNA和蛋白表达增强
TRMT10C定位在线粒体ND5 mRNA上安装m1A。这种mRNA甲基化易于抑制ND5蛋白翻译,ND5蛋白是呼吸链复合体I的一个亚基(图1A)。到目前为止,尚不清楚这种机制是否会损害线粒体的整体功能,如线粒体呼吸和膜电位,以及是否与AD等神经退行性疾病的病理变化有关。为研TRMT10C在AD中的作用,作者假设了以下实验可验证的假设。(i)TRMT10C蛋白水平在AD细胞模型和AD患者中发生改变,(ii)因此,ND5 mRNA的m1A甲基化增加,导致(iii)ND5蛋白表达减少。通过这一因果链,ND5 mRNA的m1A甲基化增强导致(iv)线粒体功能受到影响(图1A)。
为阐明第一个假设,作者使用了AD细胞模型、AD患者死后组织和公开可用的RNA-Seq数据库。研究了稳定过表达野生型人类淀粉样前体蛋白(APPwt)的HEK 293细胞中TRMT10C蛋白水平,该蛋白显示aβ1-40水平增加了10倍。在该细胞模型中,与对照细胞相比,TRMT10C蛋白水平显著增加(图1B)。在从荷兰脑库(NBB)获得的AD患者死后额叶皮层样本中,作者检测到与对照组相比TRMT10C蛋白水平显著升高(图1C)。作者只能接受13名AD患者死后组织和12名对照。此患者数量可能导致结果不足。
为进一步阐明TRMT10C在AD中的作用,作者使用了来自两个人类数据库的RNA-Seq数据。其中之一是衰老、痴呆和创伤性脑损伤(AgDemTBI)研究,分析了其中四个不同大脑区域的转录组。比较了所有未发生脑外伤和确诊AD的患者的FPKM值。与对照组相比,AD患者的TRMT10C mRNA水平没有改变(图1D)。
使用单细胞转录组学分析(scRNA-Seq)检测来自24名AD患者和24名年龄匹配的对照组额叶皮层的6种主要脑细胞类型,作者观察到兴奋性和抑制性神经元中TRMT10C mRNA水平增加。在星形胶质细胞、少突胶质细胞、前体少突胶质胶质细胞和小胶质细胞中未发现显著变化(图1E)。这种神经元特异性增加在AD的早期阶段已经很显著,并且在后期仍然存在。
图1:TRMT10C蛋白和mRNA水平在AD细胞和动物模型中以及AD患者皮层中增加
2 Aβ介导的氧化应激增强TRMT10C蛋白表达
到目前为止,作者使用Aβ过表达HEK-APPwt细胞模型、NBB-AD死后组织和数据库的数据表明,Aβ可能在TRMT10C过表达中发挥作用。然而,其他病理变化,如与氧化应激升高相关的线粒体功能障碍,也可能导致这些变化。为遵循这一想法,用低聚Aβ1-42(100 nM)或H2O2(250µM)处理HEK-Ctl细胞24小时,并分析TRMT10C蛋白的表达。在这两种应激源下,作者观察到TRMT10C蛋白表达升高(图1F)。Aß水平升高损害线粒体功能的一个众所周知的机制是通过复合物I功能障碍产生的氧化应激增加。因此,作者使用维生素C(1000µM)清除活性氧化剂。这种处理导致HEK APPwt细胞中TRMT10C蛋白表达的挽救(图1G)表明氧化应激在AD细胞模型中新描述的改变中起着至关重要的作用。
3 TRMT10C的酶伴侣亚基在AD中受到不同影响
TRMT10C需要辅因子SDR5C1在(mt)tRNA(13)的位置9上显示甲基转移酶活性;尚未研究ND5 A1374甲基化的情况是否也是如此(图2A)。TRMT10C-SDR5C1复合物也是mtRNase P的重要组成部分,需要实际核酸亚基PRORP切割。鉴于对TRMT10C的研究,这引发了一个问题,即SDR5C1和PRORP的表达在AD中是否也发生了变化。
令人惊讶的是,HEK-APPwt细胞中PRORP蛋白水平显著降低(图2B)。在AD额叶皮层死后样本中,未检测到PRORP蛋白表达的显著变化(图2C)。同样,在两个RNA-Seq数据集中也没有发现mRNA水平的差异(图2D)。
接下来,作者将重点放在SDR5C1上,发现HEK APPwt细胞中的蛋白质水平显著增强,但人类死后组织中的蛋白质含量没有显著增强(图2E,F)。AgDemTBI研究数据库显示SDR5C1 mRNA表达没有显著变化(图2G)。scRNA-Seq数据显示,SDR5C1mRNA水平仅在兴奋性和抑制性神经元中强烈上调(图2H)。
图2:WB和人类数据库评估揭示线粒体RNase P亚基SDR5C1和PRORP的不同表现
4 AD细胞模型和患者的m1A水平升高
在表明m1A写入酶TRMT10C在AD模型和人类AD皮层样本中持续上调后,作者仔细研究了一个合理的含义,即ND5 mRNA中的m1A水平是否会如假设(ii)中所指出的那样在AD病理中升高。作者首先使用基于逆转录和Illumina测序过程中m1A诱导的错误结合位点特异性分析方法分析了AD细胞模型。由于知道错误结合和逆转录阻滞的产生在很大程度上取决于反应条件和逆转录酶,作者使用了SuperScript IV(SS-IV),因为它在修饰位点上提供了高的通读能力,从而产生更适合RT-PCR和错配率分析的全长产物。作者发现,与对照相比,来自HEK APPwt细胞的总RNA样本中ND5 mRNA 1374位的错误结合水平升高(图3A),以及从线粒体提取物中分离的RNA。在这两组中,错配主要由T和G组成,这表明m1A在与SS-IV的RT过程中主要引起A和C的结合。此外,ND5 mRNA甲基化的增加反映在总RNA和线粒体RNA制剂中HEK-APPwt的跳跃率升高(图3A)。跳跃是由于m1A破坏逆转录酶而发生的核苷酸跳跃事件,是评估m1A甲基化的第二个独立参数。在NBB样本中,在第一次分析之后,m1A失配率没有改变。在作者根据Braak分期(衡量所有患者的神经病理学运动场)分离数据后,这幅景象发生了变化。当将 AD 与 Braak 阶段 5 + 6 与对照组与 Braak 阶段 0-3 进行比较时,发现错配和跳跃率显着增加(图3C)。2例AD患者携带G13708A SNP,该SNP在欧亚J单倍群中普遍存在。该SNP被证明可以防止m1A下游两个碱基的甲基化(ND5 mRNA中的1374位等于mtDNA的13710位)。在这些患者中,作者没有发现任何不匹配。因此,这些人被排除在分析之外。
生物化学证据表明,TRMT10C和SDR5C1形成一个稳定的亚复合物,作为tRNA甲基转移酶具有活性,并且能够在(mt)tRNA的第9位将腺苷和鸟嘌呤核苷酸甲基化。22 mt tRNA中的19个在第9位含有A或G。因此,作者从HEK-Ctl和HEK-APPwt细胞中分离出线粒体tRNA,并定量了m1A和m1G水平。作者没有观察到显著的变化,这表明mt tRNA已经被完全修饰,或者在与SDR5C1的复合物之外过量的TRM10C不靶向(mt)tRNA进行甲基化。
图3:ND5 mRNA 中 1374 位的 m1A 水平在 AD 模型细胞和人类患者的原代视觉皮层样本中增加
5 AD细胞模型和人类患者大脑中ND5蛋白水平降低
由于假设m1A ND5 mRNA甲基化会阻碍Watson-Crick碱基配对,(mt)tRNA在翻译过程中无法与ND5 mRNA密码子结合。结合缺陷或缺失将导致失败或至少破坏ND5蛋白的合成。然而,到目前为止,m1A甲基化与ND5蛋白水平之间的直接相关性尚未得到研究。出于这个原因,作者在AD细胞模型和AD患者的样本中进行了ND5的WB,以验证假设(iii)。
在HEK-APPwt细胞中,与对照细胞相比,ND5蛋白水平显著降低(图4A),表明m1A对ND5蛋白生物合成具有破坏性。同样,在NBB样品中,未检测到ND5蛋白水平的显著变化(图4B)。然而,与错配结果类似,当将患有Braak 5+6期的AD患者与患有Braak 0-3期的对照组进行比较时,观察到ND5的蛋白表达显著降低(图4B,C)。为研究ND5蛋白水平的改变是否可能是由于ND5 mRNA水平的降低,作者在AD细胞模型和死后组织中进行了RT-qPCR。与对照组相比,没有观察到HEK APPwt细胞的ND5 mRNA水平差异(图4D)。在死后组织中,甚至发现所有AD患者的ND5 mRNA水平都有所升高。Braak 5+6期AD患者与Braak 0-3期对照组的疗效相似(图4E,F)。
为进一步评估人类患者ND5的状态,作者使用AgDemTBI研究的数据,并观察到AD患者和健康对照组之间没有显著差异(图4G)。不幸的是,在Mathys等人的scRNA-Seq研究中没有关于ND5 mRNA的信息。
为检查ND5蛋白表达的变化是否也由Aβ1–42水平升高和氧化应激增强介导,作者用Aβ1-42(100 nM)和H2O2(250µM)处理HEK Ctl细胞24小时。ND5蛋白水平在两种应激源下均显著降低(图4H)。为评估Aβ诱导的氧化应激是否与HEK APPwt细胞中ND5蛋白表达的减少有关,再次使用维生素C(1000µM)作为抗氧化剂。在这种条件下,ND5蛋白的表达得以挽救(图4I)。
为排除Aß可能通过非特异性作用损害复合物I的所有线粒体编码亚基的翻译,在HEK-APPwt细胞中检测ND1蛋白的表达。发现HEK-Ctl细胞和HEK-APPwt细胞之间ND1蛋白表达没有显著差异。
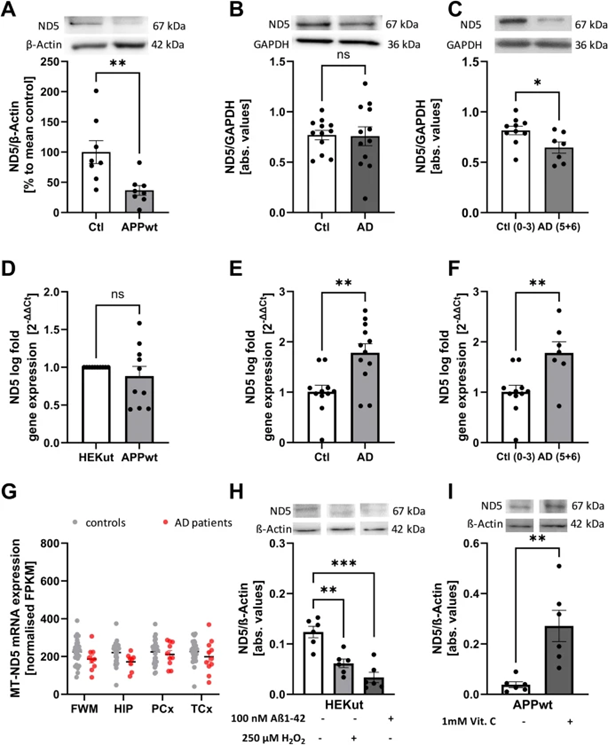
图4:ND5 蛋白水平在 AD 细胞和动物模型以及 AD 患者大脑中降低
6 TRMT10C介导的m1A甲基化与ND5蛋白翻译抑制有关
尽管在几个AD模型系统和人类患者中,TRMT10C上调,ND5 mRNA的m1A甲基化增加,ND5蛋白水平下调,但这些发现之间的假定因果关系需要进一步证实。因此,作者使用四环素诱导的过表达TRMT10C的HEK293细胞系统(pTRMT10C)来仔细研究这一假设(iv)。首先关联了24小时后0.1和1µg/mL四环素对TRMT10C蛋白表达和m1A ND5 mRNA甲基化的影响(图5A)。为排除四环素本身的影响,作者还研究了0.1和1µg/mL四环素在相应对照细胞系(Ctl)中的影响。在pTRMT10C细胞中观察到对TRMT10C蛋白表达的浓度依赖性影响,重要的是,这也与m1A mRNA甲基化相关(图5B)。
为确定mRNA甲基化增加是否导致ND5蛋白合成减少,作者使用1µg/mL四环素24小时,以诱导强大的TRMT10C蛋白过表达和相应的ND5 m1A甲基化。与用四环素孵育的对照细胞相比,pTRMT10C细胞中ND5蛋白水平显著降低(图5C)。与上述分析一致,SDR5C1和PRORP蛋白表达不受TRMT10C过表达的影响(图5D,E),ND5 mRNA表达也不受影响(数据未显示)。此外,发现TRMT10C单独增加不会导致(mt)tRNA水平的任何变化,鉴于在pTRMT10C细胞与1μg/ mL四环素孵育后,线粒体和胞质tRNA之间的比率保持不变。此外,未观察到(mt)tRNA中m1A和m1G水平的改变。
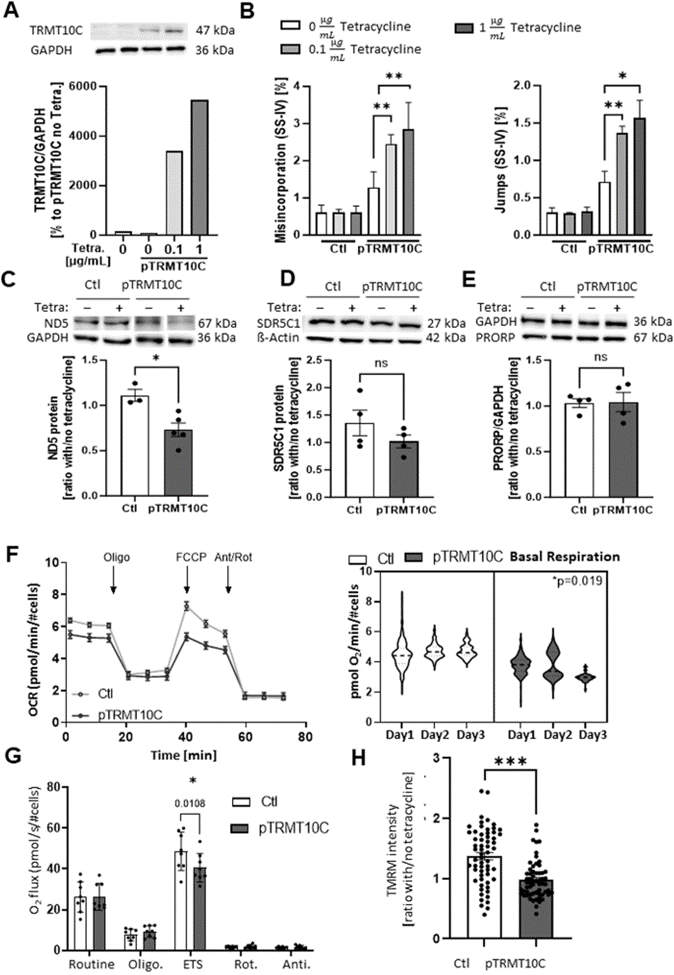
图5:四环素诱导的TRMT10C过表达及其对m1A、ND5、SDR5C1、PROP蛋白水平和线粒体功能的影响
7 TRMT10C过表达诱导严重的线粒体损伤
因为TRMT10C诱导的ND5 mRNA中的m1A甲基化降低了ND5蛋白水平,接下来,作者评估了这种降低的ND5蛋白表达是否足以诱导线粒体功能障碍。因此检测了四环素诱导TRMT10C后的线粒体功能。
首先使用Seahorse XFe 96细胞外流量分析仪,并将耗氧率(OCR)与对照细胞进行比较。首先注射寡霉素以抑制ATP合成酶,影响ATP的产生。接下来的注射是FCCP,通过ATP产生的氧摄取解耦来评估最大呼吸能力。最后,注射鱼藤酮和抗霉素来抑制复合物I和III,以估计非线粒体呼吸。代表性图表如图5F所示。值得注意的是,与同样用四环素处理的对照细胞相比,pTRMT10C细胞中的基础呼吸显著减少(图5F)。
为进一步验证这一发现,作者使用OROBOROS Oxygraph-2k进行了高分辨率呼吸测定,这证实了与对照相比,用四环素孵育的pTRMT10C细胞中的电子传输系统容量降低(图5G)。与此一致,在TRMT10C过表达的细胞中,通过TMRM染色定量的线粒体膜电位降低(图5H)。
8 功能丧失实验验证从TRMT10C到ND5蛋白表达的因果链
作者之前的实验确定了TRMT10C表达增加对mRNA甲基化、ND5水平和呼吸的影响。为了支持将这些观察结果联系起来的分子机制的假设,作者使用RNAi降低了TRMT10C水平。
与扰乱的阴性对照和未处理的HEK APPwt细胞相比,siRNA将TRMT10C蛋白表达强烈降低至30%(图6A)。这种击倒导致m1A引起的失配和跳跃率的降低(图6B,C)。ND5蛋白表达显著恢复(图6D)。作者没有观察到Aβ1-40水平的变化(图6E,F)。
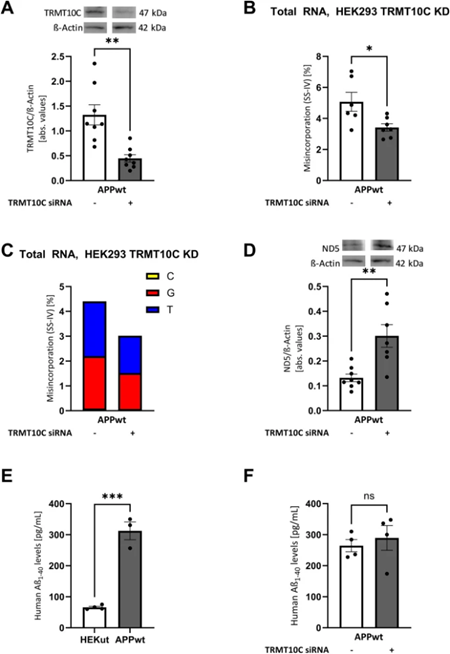
图6:siRNA诱导TRMT10C敲低及其对m1A错配和跳跃率以及mt-ND5蛋白水平的影响
结论:
作者研究了TRMT10C介导的ND5 mRNA甲基化可能导致AD复杂I功能障碍的假说。TRMT10C蛋白在AD中的表达通过ROS依赖性措施增强,导致m1A ND5 mRNA甲基化累积,从而降低ND5蛋白的表达。此外,TRMT10C蛋白表达增强会导致线粒体功能受损,这反映在线粒体呼吸和膜电位降低上。通过这项研究,作者描述了一种全新的转录后机制,即复杂I蛋白在AD中的表达和功能是如何减少的
实验方法:
细胞培养,基因敲低,ELISA,WB,RT-qPCR,线粒体功能测试,线粒体分离,线粒体tRNA分离
参考文献:
Jörg M, Plehn JE, Kristen M, Lander M, Walz L, Lietz C, Wijns J, Pichot F, Rojas-Charry L, Wirtz Martin KM, Ruffini N, Kreim N, Gerber S, Motorin Y, Endres K, Rossmanith W, Methner A, Helm M, Friedland K. N1-methylation of adenosine (m1A) in ND5 mRNA leads to complex I dysfunction in Alzheimer's disease. Mol Psychiatry. 2024 Jan 29. doi: 10.1038/s41380-024-02421-y. Epub ahead of print. PMID: 38287100.