






组蛋白乳酸化促进的ALKBH3通过SP100A的m1A去甲基化促进肿瘤进展
尽管N1-甲基腺苷(m1A)RNA修饰是RNA代谢的重要调节因子,但m1A修饰在癌变过程中的作用仍然是一个谜。在此,作者发现组蛋白乳酸化通过去除SP100A的m1A甲基化,增强ALKBH3的表达,同时减弱肿瘤抑制的早幼粒细胞白血病蛋白(PML)缩合物的形成,促进癌症的恶性转化。首先,ALKBH3在高危眼部黑色素瘤中由于组蛋白乳酰化水平过度而特异性上调,指的是m1A的低甲基化状态。此外,多组学分析随后发现,SP100A,PML小体的核心成分,可作为ALKBH3的下游候选靶点。在治疗上,ALKBH3的沉默在黑色素瘤的体内外均显示出有效的治疗效果,这可以通过消耗SP100A来逆转。在机制上,作者发现YTHDF1负责识别m1A甲基化的SP100A转录本,从而增加其RNA的稳定性和翻译效力。总之,作者最初证明了m1A修饰对于肿瘤抑制基因的表达是必要的,这扩大了目前对m1A在肿瘤进展过程中的动态功能的理解。此外,作者的研究结果表明,乳酸化驱动的ALKBH3对于PML核凝聚物的形成是至关重要的,这连接了作者对m1A修饰、代谢重编程和相分离事件的认识。该研究于2023年12月发表在《Nucleic Acids Research》,IF:14.9。
技术路线:

主要研究结果:
1 ALKBH3在眼部黑色素瘤中特异性增加并与不良后果相关
为确定m1A修饰在眼部黑色素瘤发病机制中的作用,作者首先比较了眼部黑色素瘤样本(3个CoM样本和3个UM样本)和4个对照痣样本之间的整体m1A修饰水平。值得注意的是,抗m1A斑点印迹试验显示,眼部黑色素瘤样本的m1A水平显著降低(图1A和B)。此外,去甲基化酶ALKBH3在肿瘤中蛋白表达增加(图1C-E),这与眼部黑色素瘤中m1A水平下降的结果一致。此外,ALKBH3升高与不良预后相关(图1F),进一步强调了ALKBH3在眼部黑色素瘤癌变中的重要性。
同样,在大多数黑色素瘤细胞系(MUM2B、OCM1、OMM2.3、OMM1、MEL290、92.1、92.1、CRMM1、CRMM2、CM2005.1)中,与正常色素细胞(PIG1)相比,m1A水平显著下降(图1G和H)和ALKBH3水平升高(图1I-K)。此外,高通量转录组测序进一步证实ALKBH3在眼部黑色素瘤细胞系中表达上调(图1L)。重要的是,在这些细胞中,ALKBH3与m1A水平呈显著的负相关,这表明ALKBH3的上调是导致m1A水平下降的原因(图1M)。
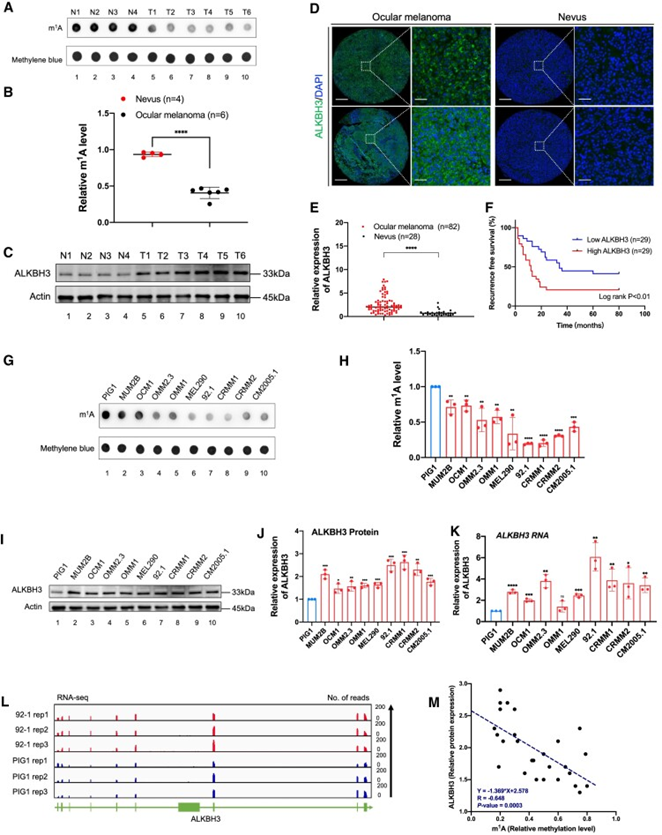
图1:眼部黑色素瘤显示ALKBH3表达增加,m1A水平降低,这与低生存率相关
2 ALKBH3在体内和体内加速眼部黑色素瘤的发生
为探索ALKBH3的致癌功能,作者使用两个单独的shRNA沉默了ALKBH3的表达(图2A和B)。同样,在ALKBH3缺陷的细胞中,观察到m1A水平显著升高(图2C)。此外,在所有测试的眼部黑色素瘤细胞中,ALKBH3沉默显著减弱细胞生长(图2D)和集落形成能力(图2E和F)。此外,Transwell实验表明,敲除ALKBH3导致迁移能力下降(图2G和H)。这些数据支持ALKBH3作为体外眼部黑色素瘤恶性增殖和转移的必要致癌因子的事实。为评估它们在体内的肿瘤形成能力,作者将对照组和alkbh3沉默的92.1黑色素瘤细胞(荧光素酶标记)注射到裸鼠中,并在原位异种移植模型中监测肿瘤生长。生物发光成像显示,ALKBH3缺陷的眼部黑色素瘤细胞的信号强度弱于对照组细胞(图2I)。此外,在ALKBH3沉默组的异种移植物的平均重量下降了约80%(图2J)。综上所述,这些实验表明,ALKBH3在眼部黑色素瘤的体内外肿瘤发生过程中都起着致癌作用。
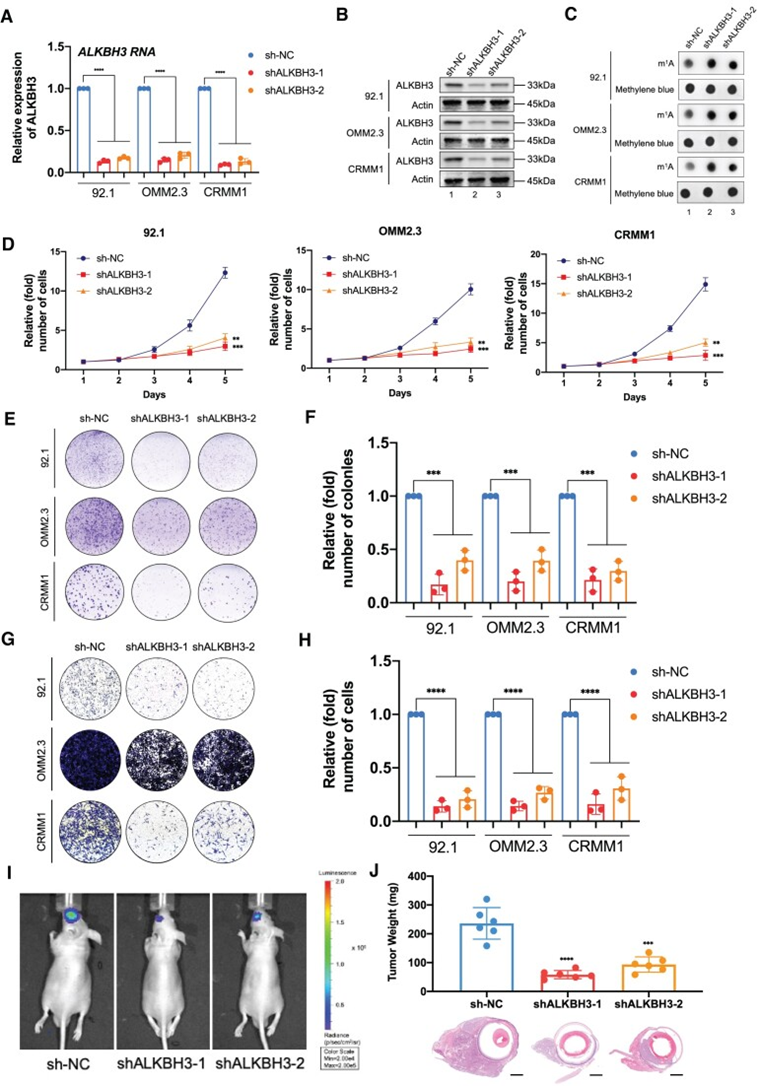
图2:ALKBH3基因的下调增加了m1A水平,并抑制眼部黑色素瘤的发生
3 组蛋白乳酸化增强了ALKBH3的过度表达
为确定ALKBH3表达增加的分子基础,作者查询了TCGA数据库,筛选了与ALKBH3共享平行表达模式的基因。根据GO和KEGG分析,作者发现ALKBH3相关基因在几个代谢方面富集,包括氧化磷酸化(P = 1.36e-05)、细胞代谢过程(P = 1.50e-05)和碳水化合物衍生物代谢过程(P = 4.18e-05),表明ALKBH3 RNA表达水平升高可能来自代谢重编程。此外,乳酸生成酶LDHA和LDHB与ALKBH3呈显著正相关,说明ALKBH3与乳酸之间呈显著相关性(图3A和B)。由于先前的研究表明组蛋白乳酸化有助于致癌基因的激活,而眼部黑色素瘤的乳酸化水平有所增加,作者假设ALKBH3水平的增加可能与组蛋白乳酸化有关。
然后,作者在眼部黑色素瘤队列中验证了泛乳酸化和组蛋白乳酸化标记物(H3K18la)之间的表达模式,这与ALKBH3蛋白的表达呈显著的正相关(图3B和C)。更重要的是,H3K18la分析的CUT&Tag和ChIPseq均显示组蛋白乳酸化信号,在ALKBH3的启动子区域捕获(存储在GEO数据库:GSE242019,图3D和E)。此外,组蛋白乳酸化抑制剂(肟酸酯和2-DG)和LDHA/B抑制均导致ALKBH3启动子区组蛋白乳酸化水平显著降低(图3F),这随后去除了所有测试的黑色素瘤细胞中ALKBH3的RNA(图3G)和蛋白水平(图3HJ)。
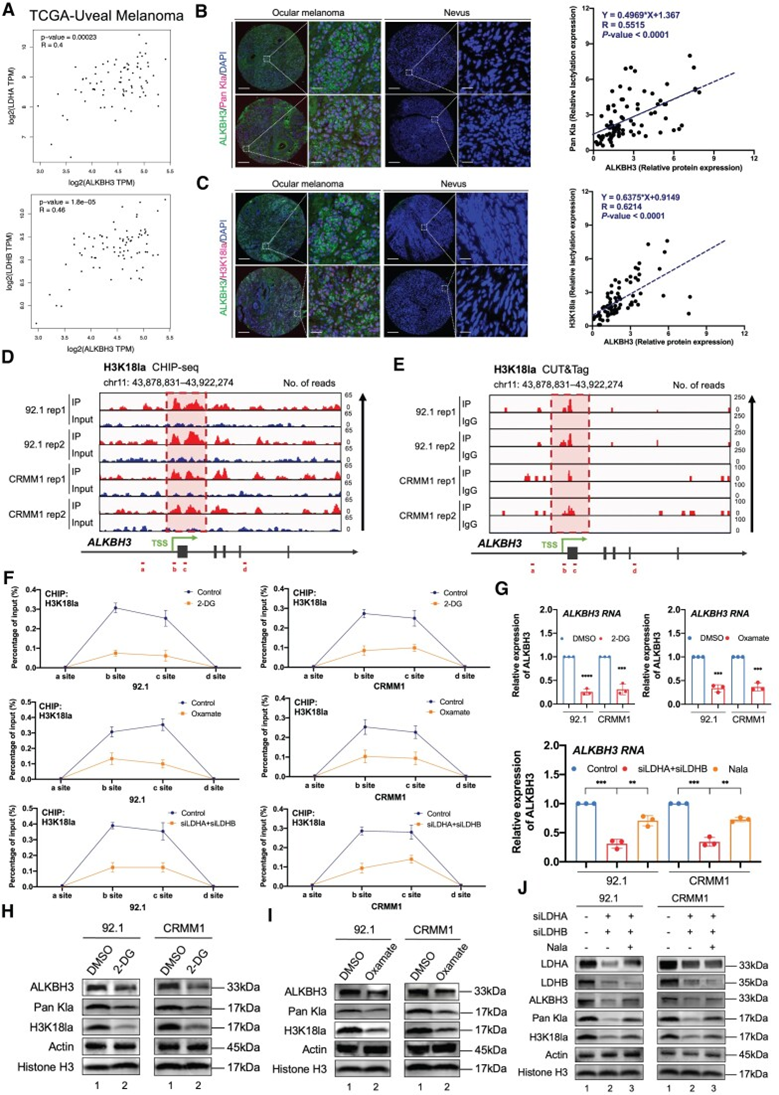
图3:组蛋白乳酸化增强ALKBH3的表达
4 多组学筛选发现SP100A为ALKBH3的下游候选基因
然后,作者探讨了ALKBH3沉默对眼部黑色素瘤细胞的抑制作用机制。由于ALKBH3负责从RNA中去除m1A修饰,作者首先对眼部黑色素瘤细胞和正常黑素细胞进行m1A-MeRIP-seq(存储在GEO数据库: GSE213748,图4A)。结果表明,从正常细胞和肿瘤细胞生成的m1A-MeRIP-seq文库中,平均鉴定出16 864个和10 212个m1A峰(图4A和B,蓝框)。与之前的m1A-meRIP-seq结果一致,m1A峰在5’UTR中富集,特别是在起始密码子附近。值得注意的是,差异表达的m1A修饰基因与多种黑色素瘤相关通路相关,包括DNA复制、mTOR/AMPK信号通路和黑色素合成,提示m1A修饰在眼部黑色素瘤的发病机制中具有调控作用。
此外,在沉默ALKBH3后,作者进行了一系列全面的高通量筛选,包括m1A-MeRIP-seq(存储在GEO数据库: GSE213748,图4C),RNA-seq(存储在GEO数据库: GSE213681,图4D和E)和对黑色素瘤细胞系的蛋白质组学分析(iTRAQ,图4F)。同样,作者注意到m1A修饰位点的变化在肿瘤相关通路中显著富集,包括PI3KAkt信号通路、局灶粘附和蛋白聚糖合成(图4C)。此外,ALKBH3的沉默导致基因表达水平的显著变化,其中有199个上调基因,74个下调基因(图4D和E,保存在GEO数据库: GSE213681)。一致地,观察到蛋白质组水平的显著变化(149个上调蛋白,67个下调蛋白,图4F),进一步强调了ALKBH3在眼部黑色素瘤发病机制中的重要性。
有趣的是,结合这些多组学数据,作者注意到在眼黑色素瘤细胞中沉默ALKBH3后,核自身抗原斑点蛋白100(SP100)的mRNA和蛋白水平均上调,随后m1A修饰水平发生显著变化(图4G-M)。值得注意的是,SP100负责PML核小体的形成,主要在各种癌症类型中作为肿瘤发生的抑制因子,包括黑色素瘤、胶质母细胞瘤、平滑肌肉瘤、乳腺癌和喉癌。这一观察结果与在ALKBH3缺陷细胞中观察到抑制效果的研究结果一致。重要的是,与正常黑素细胞相比,SP100显示眼部黑色素瘤细胞中m1A甲基化降低。有趣的是,在眼部黑色素瘤细胞中,ALKBH3抑制后,m1A修饰水平下降恢复,这可以从m1A-MeRIP-seq(保存在GEO数据库:GSE213748,图4I)和m1A-MeRIP-qPCR(图4J)中得到证明。由于最近研究发现,mRNA的m1A修饰可能增强基因的表达和翻译效果,因此ALKBH3介导的m1A修饰可能对肿瘤抑制因子SP100的表达至关重要。然后,作者验证了在ALKBH3抑制后,SP100在mRNA水平上显著上调,这可以通过RNA-seq(存储在GEO数据库中: GSE213681,图4K)和qPCR(图4L)得到证实。同样,通过iTRAQ和Western blot检测(图4M)显示,SP100在ALKBH3抑制的细胞中蛋白水平也增加。同样,在87例转移性黑色素瘤样本中,ALKBH3与SP100呈显著负相关(保存在GEO数据库中:GSE7553,R=-0.227,P=0.035)。综上所述,这些数据表明ALKBH3可能通过去除m1A修饰来抑制SP100的表达水平。
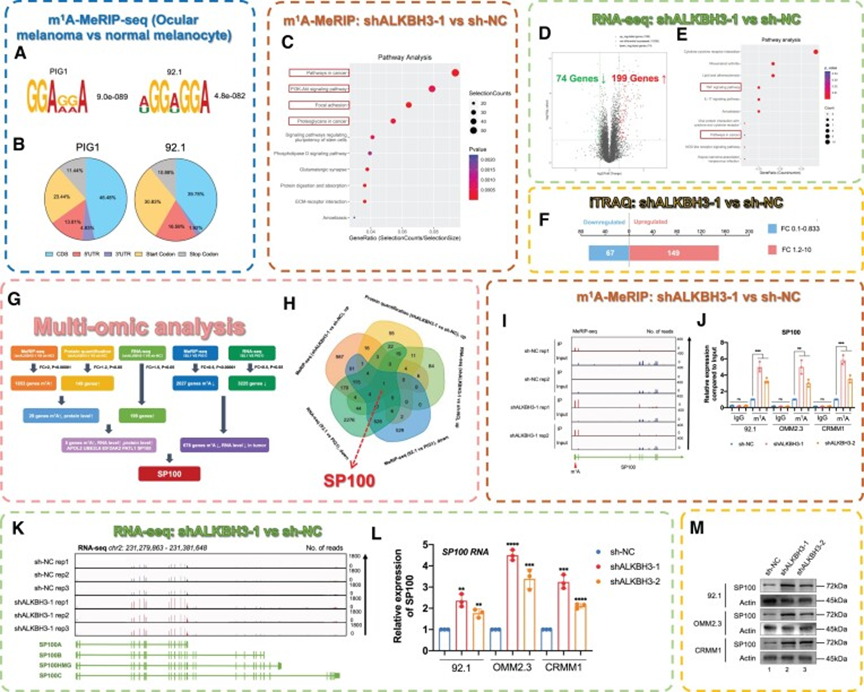
图4:ALKBH3通过去除m1A修饰来抑制SP100的表达
5 SP100A是眼部黑色素瘤中的肿瘤抑制因子
值得注意的是,在病毒感染反应的背景下,已经发现了四种显著的SP100异构型(即SP100A、SP100B、SP100C和SP100HMG),这促使作者对这些转录本的表达水平进行比较分析。值得一提的是,SP100A展现出最初15个外显子上与SP100B、SP100C和SP100和SP100HMG(1-1562bp)的共性,而最终外显子长度为395bp的差异(补充图S7A)。从眼部黑色素瘤细胞(92.1)和正常黑素细胞(PIG1)中获得的RNAseq数据显示,SP100A中有明显的信号,而SP100A中不包含的外显子中的信号可以忽略不计(图5A)。此外,作者还使用各种引物进行qPCR。这些引物包括一组专门为SP100A设计的基因(称为SP100-P1),以及另一组检测SP100B、SP100C和SP100HMG(称为SP100-P2)的基因。在眼部黑色素瘤细胞和正常色素细胞中观察到SP100A的RNA表达(图5B)。据报道,人乳腺癌细胞系ZR-75-1表达SP100HMG,作者采用ZR-75-1细胞作为阳性对照。重要的是,SP100B/SP100C/SP100HMG仅在ZR- 75-1中检测到,而在其他细胞系中未检测到。此外,这四种亚型在分子量上存在差异。全蛋白印迹显示SP100A在所有检测细胞系中(~72kDa)的特异性蛋白表达,这与之前研究中SP100A的分子量一致。研究结果表明,SP100A在我们的实验背景下大量表达,而其他亚型(SP100B、SP100C和SP100HMG)的表达可以忽略不计。
值得注意的是,与正常色素细胞相比,眼部黑色素瘤细胞中SP100A的RNA表达水平也显著下降,RNA-seq(图5A)、qPscr(图5B)和western blot检测(图5C)显示。为充分揭示SP100A在眼部黑色素瘤中的作用,作者随后检测了SP100A在眼部黑色素瘤临床样本中的表达。值得注意的是,作者发现在眼部黑色素瘤样本中,SP100A显著降低(图5D和E)。更重要的是,在作者的队列(图5F)和TCGA队列(图5G)中,SP100A的缺失与不良结果相关。
此外,根据眼部黑色素瘤样本的单细胞分析(GSE139829,使用CancerSEA平台),SP100表达与肿瘤激活特征得分降低有关,包括侵袭(R=-0.33,P<0.001)、转移(R=-0.28,P<0.001)、细胞周期激活(R=-0.19,P<0.001)、增殖(R=-0.16,P<0.001)和上皮-间充质转化(R=-0.16,P<0.001)。总的来说,这些数据表明SP100A被下调,并可能在眼部黑色素瘤中抑制一些致癌事件。
由于SP100A在眼部黑色素瘤中表达下调,作者在三种眼部黑色素瘤细胞系中外源性过表达SP100A。有趣的是,所有被检测的眼部黑色素瘤细胞在过表达SP100A后均表现出增殖能力的减弱(图5H)。此外,与对照组相比,过表达SP100A的黑色素瘤细胞形成的集落更小、更少(图5I和J)。此外,在眼部黑色素瘤细胞中引入SP100A后,可观察到其对肿瘤转移能力的显著抑制作用(图5K和L)。
最重要的是,由于SP100A作为PML小体内的分子支架,作者随后评估了PML在过表达SP100A细胞中的表达。因此,外源性过表达SP100A导致PML蛋白水平显著升高(图5M)。综上所述,这些数据进一步证实SP100A对PML的表达至关重要,这与之前的研究结果一致。此外,ALKBH3缺陷的细胞在PML小体中表现出显著升高,这与ALKBH3在调节SP100A表达过程中的关键作用相一致。综上所述,这些数据显示,SP100A负责PML核凝聚物的形成,在眼部黑色素瘤中作为肿瘤抑制因子。
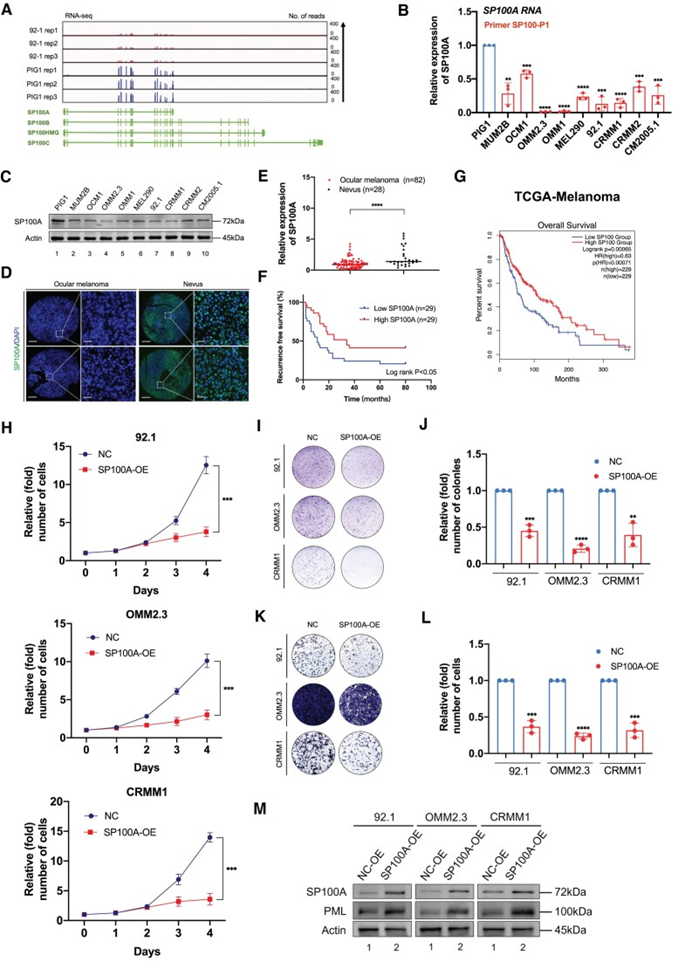
图5:SP100A在眼部黑色素瘤中起着肿瘤抑制因子的作用
6 SP100A的沉默部分损害ALKBH3缺陷细胞的肿瘤抑制效果
为进一步验证ALKBH3和SP100A表达之间的关系,作者通过转染已报道的针对SP100A的shRNA,进一步改变ALKBH3抑制后的眼黑色素瘤细胞中SP100A的表达。与预期的一样,在RNA和蛋白水平转染三个眼黑色素瘤细胞后,SP100A的表达显著降低。在细胞生长过程中,SP100A的缺失部分挽救ALKBH3抑制引导下的抑制作用(~50-60%)(图6A),而SP100A缺失的细胞对ALKBH3缺失更具有抵抗力(图6A)。SP100A沉默的细胞比对照组有更多的菌落(图6B)。此外,抑制SP100A显著增强细胞迁移能力,并削弱对ALKBH3缺陷的黑色素瘤细胞的抑制作用(图6C)。最重要的是,SP100A敲除进一步挽救ALKBH3耗竭的黑色素瘤细胞中原位肿瘤的形成(图6D和E)。同样,SP100A的稳定敲低导致野生型(图6F)和ALKBH3缺陷的眼黑色素瘤细胞(图6F)中PML的表达受损。综上所述,这些结果表明,ALKBH3通过减少SP100A介导的PML小体来促进眼部黑色素瘤。
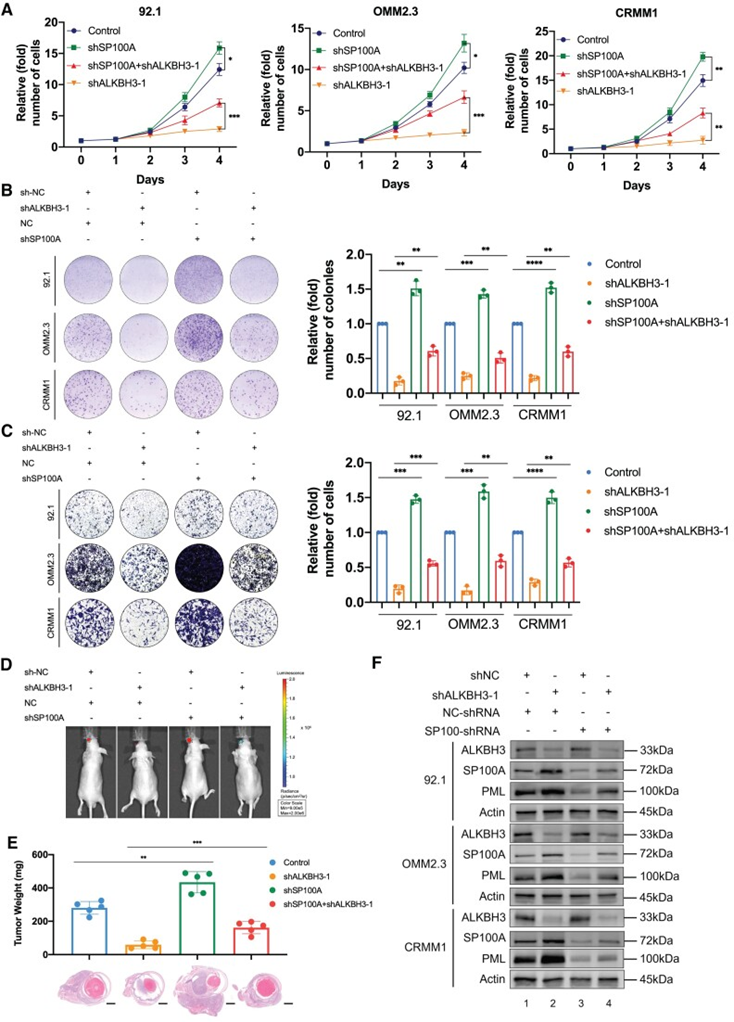
图6:SP100A沉默部分阻断ALKBH3基因下调的抗癌作用
7 SP100A的m1A修饰增强了其RNA的稳定性和翻译效力
然后,作者探讨了SP100A的m1A修饰的表观遗传机制。由于之前的研究表明YTHDF蛋白负责识别m1A甲基化,作者首先用YTHDF1、YTHDF2和YTHDF3检测SP100A mRNA的RNA binding状态。RNA免疫共沉淀分析表明,YTHDF1能特异性识别SP100A mRNA;然而,YTHDF2和YTHDF3只有有限的相互作用强度(图7A)。此外,在TCGA队列中,YTHDF1与SP100A的表达呈显著的正相关(R=0.61,P<0.0001)(图7B),这与YTHDF1是识别SP100A所必需的假设完全一致。此外,YTHDF1沉默显著抑制了SP100A的表达,并完全挽救ALKBH3沉默介导的SP100A水平的升高(图7C和D)。综上所述,YTHDF1是SP100A的解读蛋白。
然后,作者确定SP100A mRNA的特异性m1A修饰位点。在SP100A的5’UTR中,根据鉴定出的峰,作者发现5个潜在的m1A位点(图4I)[c.44A (TGTGG),c.54A(AGACG)和c.67A(TGAGG),和 c.92A/c.93A(GGAAG)]突变为T,然后将相应的野生型和突变的5’UTR克隆到pmirGLO载体中(图7E)。荧光素酶报告基因检测结果表明,c.A92T的信号下降,而其他突变组的信号保持不变(图7E)。此外,作者采用RIP-qPCR来鉴定YTHDF1与报告基因转录本(F-luc)之间的相互作用频率。观察到pmirGLOMUT4 (c.A92T)转录本在YTHDF1和F-Luc之间的结合亲和力降低,而其他转录本保持不变。这一结果与之前观察到的YTHDF1直接与m1A甲基化序列相互作用的结果一致,这被认为是RNA中m1A的“Reader”。此外,作者发现ALKBH3缺失的细胞中SP100A的RNA稳定性增强,这与ALKBH3缺失后SP100A RNA表达的增加相一致(图7F)。重要的是,在92.1细胞中进行的多聚体分析显示,ALKBH3稳定敲除导致多聚体部分的SP100A mRNA丰度显著(图7G),这通常具有有效的翻译能力。这一观察结果与之前的结论一致,即早期外显子中的m1A修饰会导致其翻译能力增强。值得注意的是,在ALKBH3基因敲低后,新生的SP100A RNA表达仍然没有发生改变。综上所述,这些结果表明,SP100A mRNA的m1A RNA甲基化有助于增加RNA的稳定性和转录后的翻译能力。
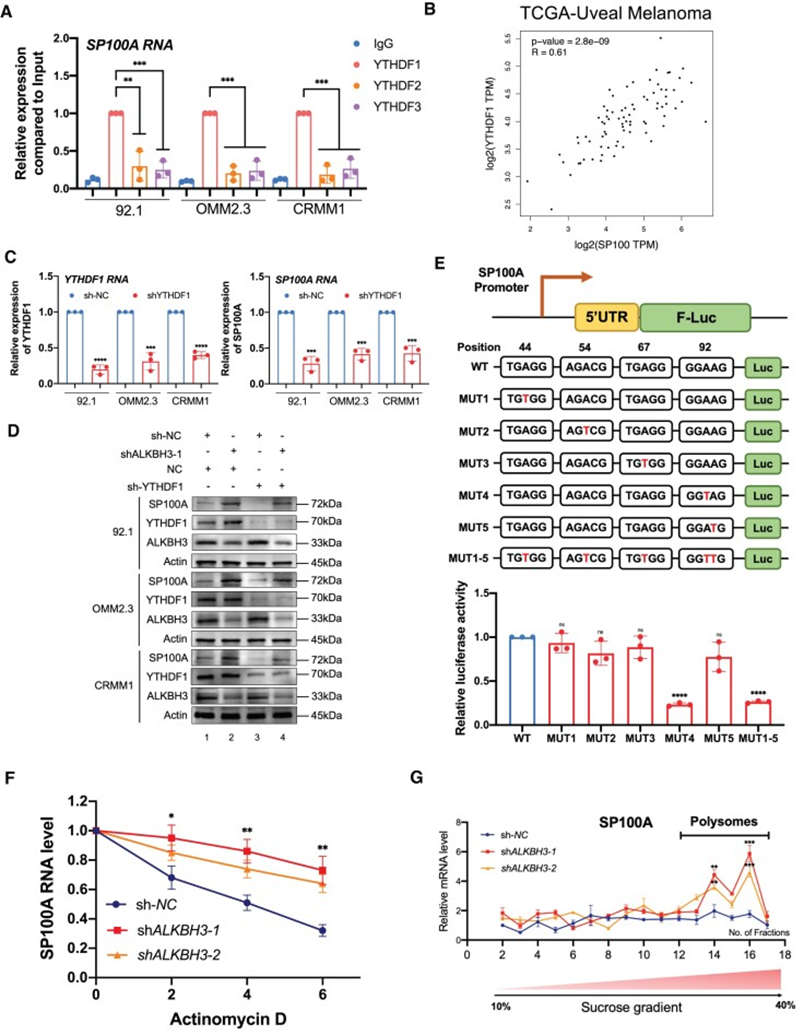
图7:SP100A的m1A修饰提高其RNA的稳定性和翻译效果
结论:
总之,作者的研究结果揭示了一个全新的肿瘤发生模型,其中m1A修饰的SP100A mRNA促进PML核缩合物的形成。此外,本研究首次揭示m1A负责肿瘤抑制基因的激活,揭示了组蛋白乳酸化、m1A修饰和相分离介导的凝聚物形成之间的串扰,从而提供了一种靶向m1A重编程的有效治疗方法。
实验方法:
细胞体外培养,斑点印迹实验,免疫荧光,WB,qPCR,质粒构建和转染,细胞增殖实验,细胞克隆实验,Transwell,异种移植物模型,ChIP-seq,CUT&Tag,MeRIP-seq,RNA-seq,RIP-qPCR,荧光素酶报告实验
参考文献:
Gu X, Zhuang A, Yu J, et al. Histone lactylation-boosted ALKBH3 potentiates tumor progression and diminished promyelocytic leukemia protein nuclear condensates by m1A demethylation of SP100A. Nucleic Acids Res. 2024;52(5):2273-2289. doi:10.1093/nar/gkad1193