






棕榈酰化允许 TNF 通路中的 RIPK1 激酶活性和细胞毒性
肿瘤坏死因子(TNF)诱导的受体相互作用丝氨酸/苏氨酸蛋白激酶 1 (RIPK1)介导的细胞死亡,包括细胞凋亡和坏死性凋亡,越来越被认为是炎症性疾病的主要驱动因素。细胞死亡检查点通常会抑制 RIPK1 激酶,以保护生物体免受其有害后果。然而,当保护性检查点被禁用时,允许 RIPK1 激酶活性的机制仍不清楚。在这里,我们确定 S-棕榈酰化是 RIPK1 激酶的许可修饰。TNF 诱导 RIPK1 棕榈酰化,由 DHHC5 介导,并依赖于 RIPK1 的 K63 连接泛素化,RIPK1 通过促进其激酶结构域的同源相互作用来增强 RIPK1 激酶活性,并在细胞死亡检查点阻断后促进细胞死亡。此外,DHHC5 在患有代谢功能障碍相关脂肪性肝炎的小鼠肝脏中被脂肪酸扩增,导致在这种情况下观察到的 RIPK1 细胞毒性增加。我们的研究结果表明,泛素化依赖性棕榈酰化使 RIPK1 激酶活性能够诱导下游细胞死亡信号传导,并表明 RIPK1 棕榈酰化是炎症性疾病的可行靶点。本文于2024年11月发表于《Molecular Cell》,IF:19.3
研究技术路线:

研究结果:
1.RIPK1在 TNF 诱导的复合物 I 中 S- 棕榈酰化
我们着手确定复合物 I 组分是否是 S- 棕榈酰化反应 TNF 感应。我们应用常用的酰基生物素交换(ABE)测定法来检测 S- 棕榈酰化。该方法涉及阻断蛋白质中的游离半胱氨酸残基,随后使用羟胺(HAM)和硫醇反应性生物素释放和捕获棕榈酰化半胱氨酸残基。我们使用基于免疫沉淀法(IP)和 ABE 分析的棕榈酰化蛋白富集,加上质谱法,来鉴定复合物 I 中的 S-棕榈酰化底物(图1A)。我们在复合物 I 中鉴定了三种酰化蛋白,包括 RIPK1,TNFR1和 TAB3(图1B)。使用2-溴棕榈酸酯(2-BP)(一种常见的 PATs 抑制剂)可以实现 S- 棕榈酰化的特异性抑制。只有 RIPK1和 TNFR1对2-BP 治疗敏感(图1C)。我们专注于 RIPK1棕榈酰化,因为 RIPK1棕榈酰化的丰度显着高于 TNFR1棕榈酰化,TNFR1棕榈酰化之前已有报道。
HEK293T 细胞中异位表达的 RIPK1被棕榈酰化,在用2-BP 处理后减少(图1D)。然后,我们使用炔标记的棕榈酸类似物15-十六烷酸(Alk14)作为代谢标记,其可以与荧光染料四甲基罗丹明(TAMRA)-叠氮化合物或通过单击化学与生物素叠氮化合物偶联。在 Alk14的代谢掺入后异位表达的 RIPK1被棕榈酰化(图1E)。相比之下,内源性 RIPK1在基础条件下不经历棕榈酰化。然而,TNF 刺激显着增加了小鼠胚胎成纤维细胞(MEFs)和骨髓来源的巨噬细胞(BMDMs)中的 RIPK1棕榈酰化(图1F)。进一步的分析显示,超过25% 的 RIPK1被棕榈酰化,如酰基聚乙二醇(PEG)交换(APE)测定所示。
我们还观察到 TNF 诱导的复合物 I 中棕榈酰化的 RIPK1显着增加,由于复合物 I10中 RIPK1的已知泛素化,其显示出高分子量的涂片模式(图1G)。RIPK1的这种修饰在2-BP 处理后减少(图1H) ,证实这些条带确实是棕榈酰化的结果。我们还观察到在 TNF 刺激的小鼠中,作为主要靶器官之一的肝组织中 RIPK1棕榈酰化的增加(图1I)。因此,RIPK1是 TNF 诱导的复合物 I 中蛋白 S- 棕榈酰化的底物。
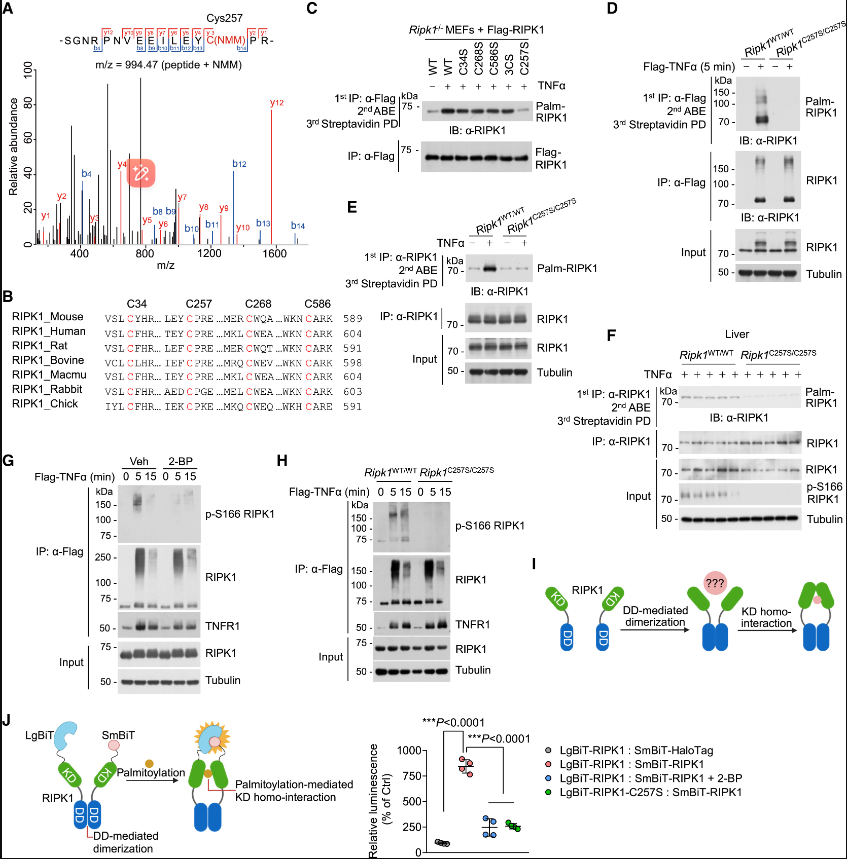
Figure 1 RIPK1在 TNF 刺激下棕榈酰化
2.RIPK1在进化上保守的 Cys257处被棕榈酰化
我们还观察到 TNF 诱导的复合物 I 中棕榈酰化的 RIPK1显着增加,由于复合物 I10中 RIPK1的已知泛素化,其显示出高分子量的涂片模式(图1G)。RIPK1的这种修饰在2-BP 处理后减少(图1H) ,证实这些条带确实是棕榈酰化的结果。我们还观察到在 TNF 刺激的小鼠中,作为主要靶器官之一的肝组织中 RIPK1棕榈酰化的增加(图1I)。因此,RIPK1是 TNF 诱导的复合物 I 中蛋白 S- 棕榈酰化的底物。
接下来,我们利用 CRISPR-Cas9技术产生 Ripk1C257S/C257S敲入小鼠。Ripk1C257S/C257S突变小鼠以正常的孟德尔比率出生,并且它们的生长看起来正常。来自 Ripk1C257S/C257S小鼠的初级 MEF 在 TNF 刺激下始终表现出有缺陷的 RIPK1棕榈酰化(图2D 和2E)。此外,与 Ripk1WT/WT 小鼠相比,Ripk1C257S/C257S小鼠的肝组织中 TNF 诱导的 RIPK1棕榈酰化降低(图2F)。我们没有观察到 WT RIPK1和 C257S 突变体之间的热稳定性或二硫键形成模式的显着差异,表明这种突变可能不会改变 RIPK1的整体折叠。
Figure 2 棕榈酰化通过增强其激酶结构域的同源相互作用来促进 RIPK1激酶活性
3.棕榈酰化促进复合物 I 中 RIPK1激酶的激活
我们接下来研究了棕榈酰化是否影响 RIPK1向复合物 I 的募集。我们对复合物 I 进行了质谱法分析,发现2-BP 不改变复合物 I 中 RIPK1的量,这通过免疫印迹证实(图2G)。与 WT RIPK1相比,RIPK1的 C257S 突变体表现出与复合物 I 相当的募集水平(图2H)。此外,2-BP 不影响复合物 I 中其他已知成分的水平。
RIPK1的激酶非依赖性激活可以通过 NF-κB 和 MAPK 途径促进细胞存活; 或者,RIPK1的激酶激活可导致细胞死亡。阻断 RIPK1棕榈酰化对 TNF 刺激的 MEF 中 NF-κB 和 MAPK 信号传导的激活没有影响。我们接下来通过在 RIPK1(p-S166 RIPK1)上使用针对磷酸化 S166的抗体来评估 RIPK1棕榈酰化是否影响其激酶活性,RIPK1是其激酶活化的公认标志物。单独的 TNF 可以激活复合物 I.41中的 RIPK1的非致死库。在2-BP 处理的 MEF 或 Ripk1C257S/C257SMEF 中,p-S166 RIPK1的水平显着降低,与各自的对照 MEF (图2G 和2H)相比,表明棕榈酰化促进复合物 I 中 RIPK1的激酶活化。鉴于 C257位于激酶结构域(KD)中,我们推测该位点的棕榈酰化可能增强 RIPK1活性。RIPK1的过度表达可导致自发激酶活化。我们发现过表达 RIPK1-C257S 突变体导致与 WT 相比 p-S166 RIPK1水平较低,并且两者都被 RIPK1激酶抑制剂 necrostatin-1s (Nec-1s)抑制。值得注意的是,C257S 突变本身不影响体外激酶测定中 RIPK1-KD 的活性,表明棕榈酰化调节 RIPK1激酶的细胞特异性机制。
4.棕榈酰化促进 RIPK1-KD 的同源相互作用
RIPK1由 N- 末端 KD,中间结构域(ID)和 C- 末端死亡结构域(DD)组成。 RIPK1的激酶活化通过二聚体内的反式自磷酸化发生,这可能是由其 KD 的同源结合介导的。众所周知,DD 介导的二聚化对于 RIPK1的自体磷酸化和激活是必不可少的。然而,KD 如何实现同源相互作用以促进自体磷酸化仍然不清楚(图2I)。我们观察到异位表达的 RIPK1-KD 的同源相互作用,其通过2-BP 处理或 C257S 突变降低,表明棕榈酰化促进 RIPK1-KD 同源相互作用。相比之下,2-BP 处理对 RIPK1-DD 的同源相互作用没有影响。我们进一步应用 NanoLuc 二元技术(NanoBiT) ,一种由结构互补的大 BiT (LgBiT)和小 BiT (SmBiT)亚基组成的双组分系统,可用于细胞内检测蛋白质相互作用,以研究棕榈酰化对 RIPK1-KD 同源相互作用的影响(图2J)。N- 末端或 C- 末端 LgBiT 融合的 RIPK1的相互作用分别用 N- 末端或 C- 末端 SmBiT 融合的 RIPK1检测,但不与阴性对照 SmBiT-HaloTag (图2J)检测,提示 RIPK1的同源相互作用。然而,2-BP 显着降低了 LgBiT-RIPK1和 SmBiT-RIPK1之间的相互作用(图2J) ,而对 RIPK1-LgBiT 和 RIPK1-SmBiT 之间的相互作用影响较小(图 S2J) ,表明 LgBiT-RIPK1和 SmBiT-RIPK1之间的相互作用信号主要来源于 RIPK1-KD 同源相互作用。此外,LgBiT-RIPK1-C257S 和 SmBiT-RIPK1之间的相互作用显着低于 LgBiT-RIPK1和 SmBiT-RIPK1之间的相互作用(图2J)。这些发现表明,在 RIPK1-DD 二聚化后,棕榈酰化对于促进 RIPK1-KD 同源相互作用是重要的,从而促进了 RIPK1的自体磷酸化。
5.当保护性检查点被禁用时,棕榈酰化使 RIPK1具有细胞毒性
我们接下来分析了 RIPK1棕榈酰化在 TNF 诱导的 RIPK1激酶介导的细胞死亡中的作用。已知有两个检查点可以抵消由 TNF 诱导的 RIPK1依赖性细胞死亡。第一个涉及通过激酶如 TGF-β 激活激酶1(TAK1) ,IκB 激酶 α/β (IKKα/β)和 TANK 结合激酶1(TBK1)磷酸化依赖性失活复合物 I 中的 RIPK1,阻止 RIPK1激酶依赖性细胞凋亡(RDA)。第二个检查点涉及复合 IIa/b 中半胱天冬酶 -8依赖性的 RIPK1切割,防止 RIPK1激酶依赖性 necroptosis。我们观察到通过 TNF 刺激联合 TAK1抑制(T/5z7) ,IKKα/β 抑制(T/TPCA-1) ,TBK1抑制(T/MRT)或 TBK1缺陷可以通过2-BP 或 RIPK1-C257S 突变。RDA 涉及 RIPK1和半胱天冬酶的激活。2-BP 处理的 MEF 或 Ripk1C257S/C257S MEF 在 T/5z7刺激下显示 RIPK1激酶活性降低和半胱天冬酶 -8介导的半胱天冬酶 -3切割(CC3)的抑制(图3C 和3D)。用2-BP 或 RIPK1-C257S 突变处理也导致复合物 IIb 的形成减少。值得注意的是,RIPK1棕榈酰化不受前 RDA 刺激的影响。TNF 诱导的促存活基因的 NF-κB 依赖性上调保护细胞免受 RIPK1激酶非依赖性细胞凋亡(RIA)。放线菌酮(CHX)可阻断 NF-κB 途径下游的翻译,从而促进 RIA,其不能被 Nec-1s 阻断。2-BP 或 RIPK1-C257S 突变对 TNF/CHX 诱导的 MEF 死亡没有影响,表明棕榈酰化在 RIA 中不起作用。
使用抑制剂如Z-Val-Ala-Asp-FMK (zVAD)抑制 caspase-8可诱导 TNF 刺激的细胞中 RIPK1激酶依赖性坏死。用 CHX 和 zVAD (T/C/Z)联合治疗 TNF 是可被 Nec-1s38,43抑制的 necroptosis 的完善模型(图3E)。我们发现2-BP 或 RIPK1-C257S 突变显着降低了细胞对 T/C/Z 诱导的 necroptosis 的敏感性(图3E 和3F)。坏死也发生在用 TNF 和 zVAD 处理的细胞中,伴随5z7(T/5z7/Z)抑制 TAK1,50也被2-BP 或 RIPK1-C257S 突变减少。此外,2-BP 处理的细胞或 Ripk1C257S/C257S 细胞对 necroptosis 诱导的敏感性降低通过 p-S166 RIPK1和坏死生物标志物 p-T231/S232 RIPK3和 p-S345 MLKL20,51(图3G,3H)的降低进一步标志。最后,2-BP 或 RIPK1-C257S 突变导致坏死体形成减少。值得注意的是,RIPK1棕榈酰化不受促坏死触发因子的影响。RIPK1激酶依赖性细胞死亡严重参与介导 TNF 诱导的系统性炎症反应综合征(SIRS)。2-BP 或 RIPK1-C257S 敲入突变有效地保护小鼠免受致死剂量的 TNF 诱导的致死性(图3I 和3J) ,进一步支持棕榈酰化在促进 RIPK1细胞毒性中的作用。
当巨噬细胞中半胱天冬酶 -8被抑制时,RIPk1介导的 necroptosis 也可以在 Toll样受体(LPS)刺激的脂多糖4(TLR4)途径中触发。引人注目的是,与相应的对照细胞相比,2-BP 处理的 BMDMs 或 Ripk1C257S/C257S BMDMs 显示对由 LPS 联合 zVAD (LPS/Z)诱导的 necroptosis 的敏感性降低。用 LPS/Z 刺激的 BMDM 中的2-BP 处理也降低了 p-S166 RIPK1,p-T231/S232 RIPK3和 p-S345 MLKL 的水平。总的来说,这些结果表明,RIPK1棕榈酰化许可其激酶活性和细胞毒性失活的细胞死亡检查点。
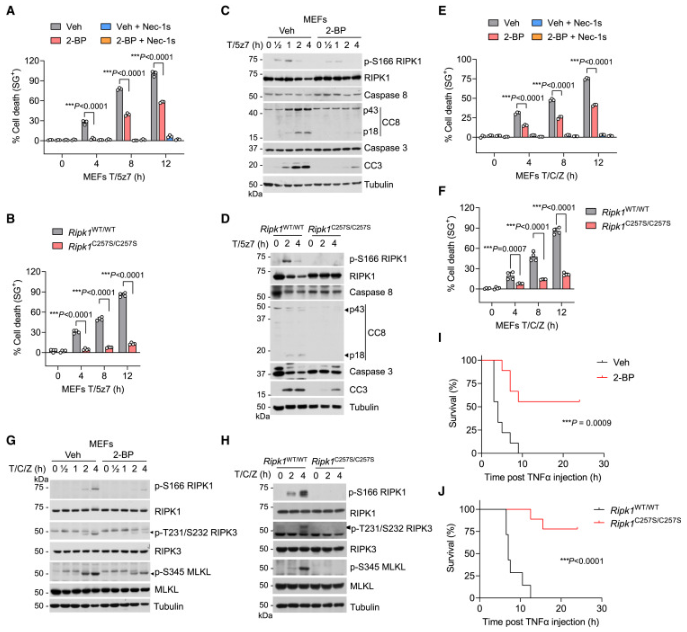
Figure 3 棕榈酰化促进 RIPK1对细胞死亡检查点的细胞毒性阻断
6.DHHC5介导复合物 I 的 RIPK1棕榈酰化
在哺乳动物中,DHHC 家族由23个成员组成。我们使用短发夹 RNA (shRNA)敲低这23种 DHHC 蛋白在 MEF 中的表达,并鉴定了6种在敲低时抑制 RDA 和/或 necroptosis 的 PATs (Z 评分 > 1)(图4A)。RIPK1与六个预选的 PATs 中的每一个的过度表达确定只有 DHHC5增强 RIPK1棕榈酰化。此外,DHHC5的缺失大大降低了异位表达的 RIPK1的棕榈酰化。
在 DHHC5敲低的细胞中,但在其他 DHHC 敲低的细胞中,TNF 诱导的 RIPK1棕榈酰化被消除。我们进一步通过在炔烃标记的棕榈酰辅酶 A (CoA)(Alk14-CoA)存在下将纯化的 RIPK1蛋白与 DHHC5温育来进行体外棕榈酰化测定。我们检测了由 DHHC5催化的 RIPK1的体外棕榈酰化,但不是通过在保守的 DHHC 基序(DHHS5)31(图4C)内含有 Cys-to-Ser 取代(C134S)的 DHHC5的催化失活突变体。当在体外棕榈酰化测定中与 DHHC5和 Alk14-CoA 温育时,RIPK1-C257S 突变体也表现出棕榈酰化缺陷(图4D)。与 WT 细胞相比,我们观察到 DHHC5缺陷型细胞中复合物 I 中棕榈酰化的 RIPK1显着减少(图4E) ,表明 DHHC5对于 TNF 诱导的复合物 I 中的 RIPK1棕榈酰化是必需的。
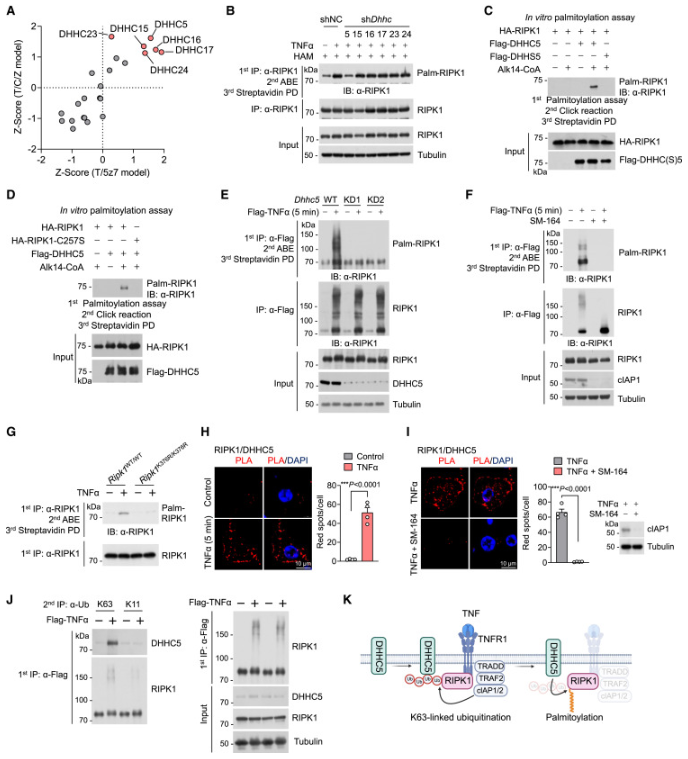
Figure 4 DHHC5介导 TNF 诱导的复合物 I 中 RIPK1棕榈酰化
7.DHHC5介导的棕榈酰化依赖于 K63连接的 RIPK1泛素化
RIPK1在复合物 I 中高度泛素化,作为多种蛋白质复合物募集的支架。我们推测抑制 RIPK1泛素化是否可能影响其棕榈酰化。在 TNF 感应后,RIPK1和其他复合物 I 组分迅速经历由 cIAP1/2介导的 K63和 K11连接的泛素化以及由线性泛素链组装复合物(LUBAC,包含 HOIL-1,HOIP 和 SHARPIN)介导的线性泛素化。值得注意的是,复合物 I 中 RIPK1的棕榈酰化在 SM-164处理的 cIAP1/2消耗后被消除(图4F)。然而,其中 LUBAC 功能失调的 Hoip-/-MEF 表现出与 WT MEF 相似程度的 RIPK1棕榈酰化。这些结果表明 RIPK1棕榈酰化依赖于 cIAP1/2介导的泛素化。与此相一致的是,2-BP 对 TNF 联合 SM-164(T/S)或 T/S/Z 诱导的 necroptosis 诱导的 RDA 没有影响。相比之下,已知 HOIP 缺陷使细胞对 RIPK1激酶依赖性细胞死亡敏感,但2-BP 仍然在 HOIP 缺陷型 MEF 中提供了对 RDA 和 necroptosis 的保护。CIAP1/2介导的 RIPK1的泛素化主要发生在复合物 I中的鼠 RIPK1(人 RIPK1的 K377)的 Lys376(K376)上。值得注意的是,表达 RIPK1-K376R 突变体的细胞显示消除的棕榈酰化(图4G) ,并且它们在 RDA 和 necroptosis 方面对2-BP 处理没有反应。
我们观察到在 TNF 处理的 MEF 中,RIPK1和 DHHC5之间的相互作用显著增加,如通过邻近连接测定(PLA)所确定的(图4H)。因此,DHHC5和 RIPK1被招募到复合物 I 中。然而,在用 SM-164处理的细胞中,RIPK1和 DHHC5之间的 TNF 诱导的相互作用完全消除(图4I) ,表明在 RIPK1上加入 cIAP1/2的泛素链促进 DHHC5的募集。CIAP1/2诱导 RIPK1的 K63和 K11连接的泛素化。我们发现 DHHC5向复合物 I 的募集主要由 K63连接的泛素链介导(图4J)。因此,cIAP1/2介导的 K63连接的 RIPK1泛素化促进 DHHC5募集至复合物 I,允许复合物 I 中 DHHC5介导的 RIPK1的棕榈酰化(图4K)。
8.DHHC5促进 TNF 诱导的 RIPK1激酶活化和细胞毒性
随后我们研究了 DHHC5在促进 RIPK1激酶活化和细胞毒性中的作用。当细胞接受 TNF 刺激时,DHHC5缺陷导致复合物 I 中 p-S166 RIPK1的减少(图5A)。结果,DHHC5缺陷细胞对 T/5z7诱导的 RDA 的敏感性降低(图5B)。此外,DHHC5缺陷显着降低了用 T/5z7处理的细胞中的 RIPK1和半胱天冬酶活化以及复杂的 IIb 形成(图5C)。同样,DHHC5缺陷细胞对 T/C/Z 诱导的坏死的敏感性显著降低(图5D)。一致的是,DHHC5缺陷导致用 T/C/Z 处理的细胞中 p-S166 RIPK1,p-T231/S232 RIPK3,p-S345 MLKL 和坏死体形成的减少(图5E)。与棕榈酰化在促进 RIPK1-KD 同源相互作用中的作用一致,DHHC5缺陷降低了 RIPK1的 KD-KD 相互作用(图5F)。
综上所述,这些结果建立了以下模型,说明 RIPK1激酶活化是如何被许可促进细胞毒性的(图5G)。在 TNF 感应后,RIPK1被迅速募集到复合物 I 中,在那里它经历 cIAP1/2介导的 K63连接的泛素化,其促进 DHHC5的募集,其催化 RIPK1-KD 的棕榈酰化。当细胞死亡检查点被阻断时,棕榈酰化促进 RIPK1-KD 的同源相互作用并增强 RIPK1反式激活,最终导致细胞死亡。
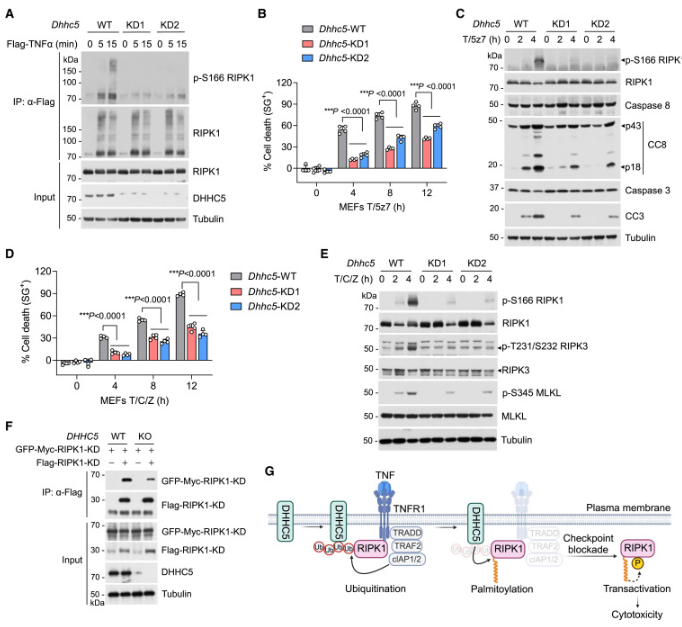
Figure 5 DHHC5在细胞死亡检查点阻断时促进 RIPK1激酶活性和细胞毒性
9.DHHC5在 MASH 肝脏中通过脂肪酸扩增
以前的研究已经将 RIPK1的细胞毒性与 MASH 联系起来,其中肝脏积聚了过多的脂肪沉积物。然而,MASH 中 RIPK1激活的机制仍然难以捉摸。我们发现喂食胆碱缺陷型高脂饮食(CD-HFD)的小鼠可以迅速诱导 MASH 组织病理学,与正常脂肪饮食(ND)的小鼠相比,RIPK1棕榈酰化显着增加(图6A)。与喂食 ND 的小鼠相比,喂食 CD-HFD 或常用 HFD 的小鼠的肝组织中 DHHC5的蛋白质水平也升高(图6B)。此外,与喂食 ND 的小鼠相比,喂食 CD-HFD 的小鼠的肝组织中 Dhhc5的 mRNA 水平显着升高。
我们在 MASH 肝脏中观察到 DHHC5在肝细胞中的特异性上调,但在非实质(NPC)细胞中没有(图6C)。然后,我们用0.4 mM 棕榈酸(PA)刺激原代肝细胞,在该浓度下进行以模拟脂肪肝疾病的体内条件。我们观察到 PA 处理后肝细胞中 Dhc5 mRNA 和蛋白质水平的增加(图6D)。此外,在 PA 处理的肝细胞中 TNF 诱导的 RIPK1棕榈酰化增加。Dhc5的启动子区域含有转录因子 c-jun66的结合位点(图 S6F)。已经证实,PA 激活 c-Jun N- 末端激酶(JNKs) ,其磷酸化 c-Jun 并促进 MASH。我们假设 PA 诱导的 DHHC5的转录激活是由 JNK/c-Jun 途径介导的。一致认为,JNK 的药理学抑制阻止了 PA 处理的肝细胞中 DHHC5的增加(图6D)。细胞外信号调节激酶(ERK)信号传导和 p38信号传导在 MASH 肝脏和 PA 刺激的肝细胞中也被激活,这促进了 c-Jun 驱动的基因转录。一致地,抑制 ERK 或 p38信号传导也降低了 PA 诱导的 DHHC5的表达。
我们通过使用被 JNK 抑制消除的染色质免疫沉淀定量实时 PCR (ChIP-qPCR)观察到 PA 处理的肝细胞中 DHHC5启动子处 c-Jun 的强占据(图6E)。然后,我们通过将 Dhhc5启动子区与荧光素酶基因连接进行荧光素酶报告基因检测(图6F)。C-Jun 的过表达显着增加了通过 Dhhc5启动子的转录激活,但不是通过对照 CMV 启动子或缺乏 c-Jun 结合位点的突变的 Dhhc5启动子(图6F)。因此,DHHC5通过 JNK/c-Jun 途径在 MASH 肝细胞中被脂肪酸扩增。

Figure 6 脂肪酸扩增的 DHHC5促进 MASH 中 RIPK1驱动的肝损伤
10.脂肪酸扩增的 DHHC5促进 MASH 中 RIPK1驱动的肝损伤
接下来,我们通过将 Dhhc5f/f 小鼠与白蛋白 -Cre (Alb-Cre)转基因小鼠杂交,产生肝细胞特异性 Dhhc5敲除小鼠,并用 CD-HFD 喂养小鼠以诱导 MASH 相关的组织病理学。在来自 CD-HFD 喂养的小鼠的原代肝细胞中,DHHC5缺陷显着降低了棕榈酰化 RIPK1的水平(图6G)。与以前的研究一致,我们观察到通过 p-S166 RIPK1免疫染色确定的 RIPK1激酶活性增加,并且通过末端脱氧核苷酸转移酶介导的脱氧尿苷三磷酸缺口末端标记(TUNEL)染色观察到细胞死亡,在 CD-HFD 喂养的小鼠的肝切片中,与 ND 喂养的小鼠相比。CD-HFD 喂养的 Dhhc5f/f; Alb-Cre 小鼠的肝脏中 p-S166 RIPK1和 TUNEL 阳性细胞的量减少(图6H)。与这一发现一致,在 CD-HFD 喂养的 dhc5f/f; Alb-Cre 小鼠(图6I)中,血清谷丙转氨酶(ALT)和天冬氨酸转氨酶(AST)水平显着降低,这是肝损伤的两个常见标志物。
苏木精和伊红(H & E)和油红 O (ORO)染色显示,与 Dhhc5f/f 小鼠相比,CD-HFD 喂养的 Dhc5f/f; Alb-Cre 小鼠的肝脂肪变性和脂质积累没有改变。这一发现与之前的研究一致,表明抑制 RIPK1激酶不影响 CD-HFD 模型中的肝脂肪变性,其刺激严重的 MASH 病理学。相比之下,我们观察到 CD-HFD 喂养的 Dhhc5f/f; Alb-Cre 小鼠与 Dhc5f/f 小鼠相比,通过染色巨噬细胞标志物 CD45(图6J)测量。DHHC5缺陷也导致促炎细胞因子 Tnf 和 Il6以及趋化性细胞因子 Ccl2,Ccl5和 Cxcl1的下调(图6K)。此外,与对照小鼠相比,通过 Masson 三色染色(MTS)和天狼星红染色评估的肝纤维化在 CD-HFD 喂养的 Dhc5f/f; Alb-Cre 小鼠中减少(图6L)。与纤维化减少一致,dhHC5缺乏下调胶原基因 Col1a1,纤维化生长因子 Pdgfa,Pdgfb 和受体 Pdgfra 的表达(图6M)。当用 Nec-1s 处理这些小鼠时,DHHC5缺陷对 CD-HFD 喂养的小鼠的肝损伤的影响受到损害(图6N) ,表明 DHHC5通过 RIPK1激酶依赖性方式在促进 MASH 中的肝损伤中起作用。
11.缺陷型 RIPK1棕榈酰化改善 MASH 肝损伤
与 DHHC5缺陷小鼠类似,与 Ripk1WT/WT 小鼠相比,CD-HFD 喂养的 Ripk1C257S/C257S小鼠的肝脏中的肝脂肪变性没有改变。然而,在 CD-HFD 喂养的 Ripk1C257S/C257S小鼠的肝脏中,RIPK1激活和细胞死亡被有效抑制(图7A)。因此,在 CD-HFD 喂养后,与 WT 小鼠相比,Ripk1C257S/C257S小鼠血清 ALT 和 AST 水平显著降低(图7B)。RIPK1-C257S 突变也导致炎症反应显着降低,如 CD-HFD 喂养的小鼠肝脏中巨噬细胞浸润减少和细胞因子和趋化因子下调所示(图7C 和7D)。此外,CD-HFD 喂养后,组织学纤维化显着降低,Ripk1C257S/C257S小鼠的肝脏中的纤维化基因谱紊乱也得到改善(图7E 和7F)。此外,当用 Nec-1s 处理这些小鼠时,RIPK1-C257S 突变对 CD-HFD 喂养的小鼠的肝损伤的影响受到损害(图7G) ,进一步表明 RIPK1棕榈酰化在促进体内 RIPK1细胞毒性中的重要性。
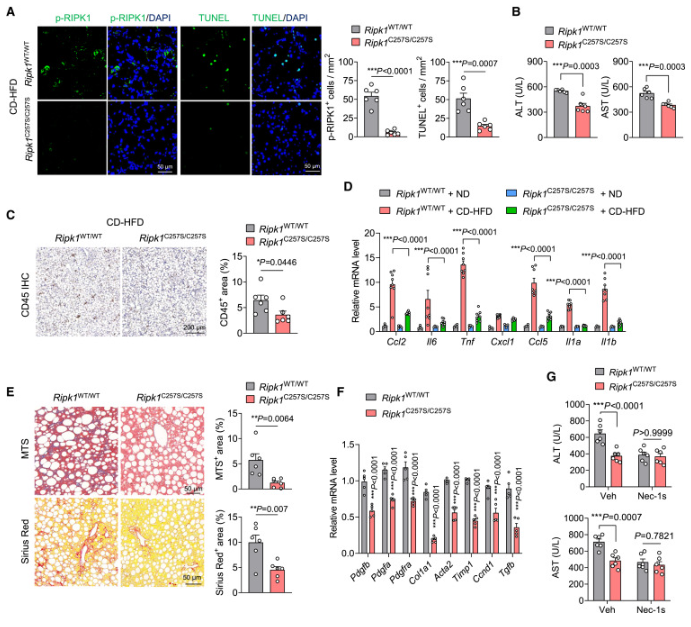
Figure 7 RIPK1棕榈酰化缺陷减轻 MASH 相关性肝损伤
12.靶向 DHHC5对小鼠 MASH 的治疗作用
我们接下来评估了靶向 DHHC5对 MASH 相关性肝损伤小鼠的潜在治疗作用。我们产生了编码靶向肝靶向治疗基因载体 DHHC5(71)的短发夹 microRNA 的腺相关病毒8(AAV8) ,以诱导 MASH 肝细胞中的 DHHC5敲低。我们用 CD-HFD 喂养小鼠4周以建立 MASH,然后静脉注射 AAV-shDhc5的小鼠,同时继续 CD-HFD 4周。在 AAV 感染4周后,我们在 AAV-shDhc5小鼠的肝脏中检测到大量的 DHHC5减少。DHHC5敲低不影响 CD-HFD 喂养的小鼠的肝脂肪变性。尽管如此,DHHC5敲低显著降低了 CD-HFD 喂养小鼠肝切片中 RIPK1的活化和细胞死亡。此外,与 AAV-shNC 小鼠相比,AAV-shDhc5小鼠血清 ALT 和 AST 水平显著降低。DHHC5敲低也减轻了巨噬细胞浸润和肝脏炎症。此外,AAV-shDhhc5注射液显着抑制组织学肝纤维化和促纤维化标志物的表达。这些结果表明,靶向 DHHC5可以减轻 MASH 相关的肝损伤。
研究结论:
因此,预防肝损伤和肝纤维化是 MASH 治疗的一个主要目标,可以通过改善肝脏死亡来实现。因此,我们的研究具有潜在的临床意义,并提出 DHHC5和 RIPK1棕榈酰化作为更具体治疗 MASH 相关性肝损伤的可行靶点。
实验方法
基因表达数据分析、启动子序列分析、基因编辑与突变、体外激酶检测、荧光素酶报告基因实验、免疫共沉淀(IP)和免疫印迹(IB)、细胞培养与处理、细胞毒性分析、蛋白-蛋白相互作用检测、代谢标记与荧光检测、免疫荧光与共定位分析、小鼠模型、组织学染色评估肝脂肪变性和纤维化、检测血清中肝损伤标志物水平
参考文献:
Zhang N, Liu J, Guo R, Yan L, Yang Y, Shi C, Zhang M, Shan B, Li W, Gu J, Xu D. Palmitoylation licenses RIPK1 kinase activity and cytotoxicity in the TNF pathway. Mol Cell. 2024 Nov 21;84(22):4419-4435.e10. doi: 10.1016/j.molcel.2024.10.002. Epub 2024 Oct 28. PMID: 39471814.