






三阴性乳腺癌的治疗新思路:PEAK1与CAMK2的信号调控与药物干预
PEAK假激酶家族,包括PEAK-1-3,在几种预后不良的人类癌症中发挥致癌作用,包括三阴性乳腺癌(TNBC)。然而,由于假激酶缺乏催化活性,治疗靶向是具有挑战性的。为了解决这个问题,我们筛选了PEAK1效应物,并鉴定了钙/钙调素依赖性蛋白激酶2(CAMK2)D和CAMK2G。PEAK1通过PLC-γ- 1/Ca2+信号传导和直接结合CAMK2促进TNBC细胞中CAMK2的激活。反过来,CAMK2磷酸化PEAK1以增强与PEAK2的关联,这对PEAK1的致癌信号传导至关重要。为了实现PEAK1/CAMK2的药理学靶向,我们重新使用了第二代CAMK2抑制剂RA306。RA306抑制PEAK1增强的TNBC细胞在体外的迁移和侵袭,并显著减弱TNBC异种移植物的生长和转移,其方式与PEAK1消融相一致。总的来说,这些研究确定了PEAK1是一个关键的细胞信号传导纽带,整合Ca2+和酪氨酸激酶信号,并确定CAMK2是PEAK1下游的治疗“可操作”靶标。
机制示意图:
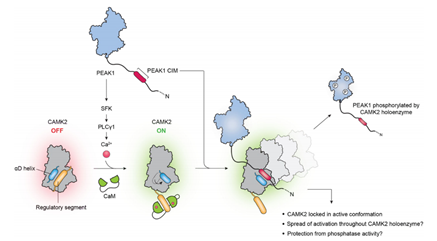
主要研究结果:
1. 用BiCAP表征PEAK1和PEAK2二聚体特异性相互作用组
为了表征PEAK1和PEAK2同型二聚体的相互作用组,以及PEAK1/PEAK2异源二聚体,我们应用了双分子互补亲和纯化结合串联质谱(BiCAP-MS/MS)。特异性二聚体复合物被亲和纯化,并通过LC-MS/MS以数据独立的获取模式鉴定相关蛋白(图1A-C)。Grb2是一种特性良好的PEAK1结合了PEAK1同型二聚体和PEAK1/PEAK2异源二聚体,但不结合PEAK2同型二聚体,这与我们之前的结果一致,PEAK1将Grb2与PEAK216连接起来。两个蛋白磷酸酶1 (PP1)家族成员PPP1CA和PPP1CC先前在传统的基于免疫亲和力的MS/MS筛选中被鉴定为PEAK1相互作用物。然而,它们对PEAK1和PEAK2的选择性尚不清楚。在这里,与Grb2一样,它们被鉴定为PEAK1同型二聚体和PEAK1/PEAK2异源二聚体的相互作用物,PP1家族的另一个成员PPP1CB与PEAK1同型二聚体相关。最近的一篇论文报道了14-3-3蛋白在调节PEAK3效应募集和生物活性方面的关键调控作用,多个14-3-3亚型结合了同型二聚体和异源二聚体。进一步鉴定的相互作用物包括RAC1鸟嘌呤核苷酸交换因子(GEF) FARP1和MAPK途径拮抗剂SPRY4,它们对PEAK2同型二聚体具有选择性。此外,Ca2+/钙调素依赖性蛋白激酶2 (CAMK2)D和G与PEAK1和PEAK2同型二聚体以及PEAK1/PEAK2异源二聚体相关,而CAMK2B则被PEAK2同型二聚体和PEAK1/PEAK2异源二聚体募集(图1A-C)。对每个PEAK复合物的相互作用组进行生物信息学分析,以确定富集的途径/过程,确定了包括CAMK2酶在内的钙相关途径被富集(特别是PEAK1和PEAK2同型复合物的“突触后信号传导”,以及所有3种复合物的“肌层”和“心脏信号传导”)。
总的来说,这些数据为PEAK1/2复合物的结合选择性提供了重要的见解,并确定了特定的CAMK2家族成员作为这些假激酶支架的相互作用物。
2. CAMK2与PEAK1和PEAK2相互作用的表征
为了确认特定CAMK2家族成员与PEAK1和PEAK2的关联,我们将重点放在CAMK2D和CAMK2G上,因为这些激酶与BiCAP-MS/MS分析的所有同型和异型PEAK复合物相关。这些CAMK2s与内源性PEAK1和PEAK2的相互作用在MDA-MB-231乳腺癌细胞中通过近端结扎试验进行了研究(图1D-F)。使用这种技术,可以检测到PEAK1与CAMK2D或G之间的相互作用,并且当使用siRNA敲除PEAK1时,PEAK1与CAMK2D或G之间的相互作用显著减少(图1D-E)。PEAK2与CAMK2D或G之间的相互作用也观察到类似的结果(图1D-F)。这些数据表明,在体内,CAMK2D和G在内源性表达水平上与PEAK1和PEAK2相互作用。
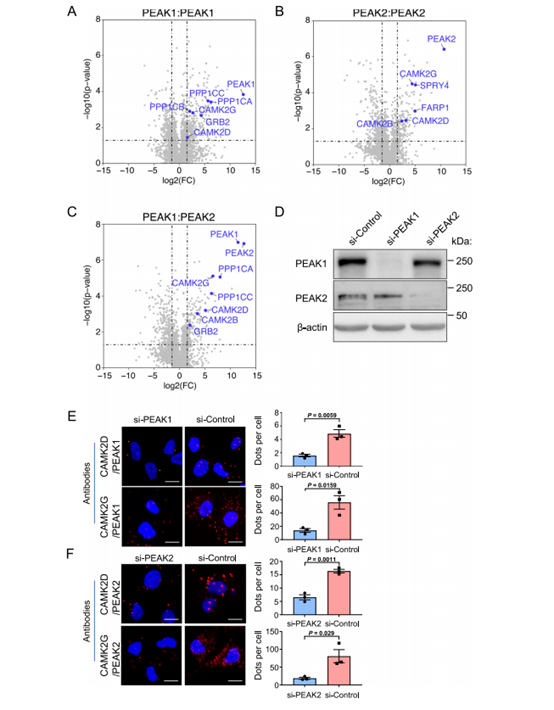
图1 通过BiCAP-MS/MS鉴定特定CAMK2亚型为peak - 1/2相互作用物
3. PEAK1对CAMK2活性的调控
已知的PEAK1在乳腺癌中的过表达,使我们质疑PEAK1/CAMK2相互作用的功能后果。随着细胞内Ca2+的增加,Ca2+/calmodulin (CaM)与CAMK2调控片段的结合导致调控片段从激酶结构域释放、激酶激活和T286的磷酸化(CAMK2D和G中的T287)。在T286磷酸化存在的情况下,CAMK2调控片段不能再介导自抑制,CAMK2的活性是Ca2+/CaM自主的。在MDA-MB-231 TNBC细胞中,PEAK1的敲低不影响CAMK2D和G的表达水平,但显著降低了它们的激活,这是通过T287位点的磷酸化确定的(图2A,B)。此外,这些细胞中PEAK1的过表达显著增强了这两种CAMK2亚型的激活(图2C,D)。在MDA-MB-468 TNBC细胞中,也观察到PEAK1对CAMK2D/G活性的正向调节(补充图2)。一种膜透性Ca2+螯合剂阻断Ca2+对CaM的激活,显著降低了CAMK2 T287的基础磷酸化,并完全阻断了PEAK1对CAMK2激活的作用,表明PEAK1介导的CAMK2激活依赖于Ca2+(图2E,F)。
根据这一结果,我们确定PEAK1是否可以调节Ca2+水平,从而间接调节CAMK2的激活。Fluo-4成像的应用显示,在稳态下,PEAK1过表达显著增强了细胞内Ca2+水平(图2G,H)。关于PEAK1调节Ca2+信号传导的机制,已知的峰作为特定酪氨酸激酶的调节剂和/或底物的作用,使我们确定PEAK1过表达是否影响酪氨酸磷酸化,从而激活PLC- γ1。导致IP3的产生,从而触发细胞内储存的Ca2+释放。事实上,PEAK1敲低降低了PLCγ-1 Y783的酪氨酸磷酸化(图2I),而PEAK1过表达增强了该位点的PLCγ-1酪氨酸磷酸化(图2J)。然而,鉴于PEAK1是一种假激酶,PLC γ-1的磷酸化必须由PEAK1下游的真正酪氨酸激酶介导。由于PEAK1是TNBC27中一个突出的Src家族激酶(SFK)信号网络的成员,而PLCγ1是SFKs28的已知底物,我们假设SFK可能介导了这一作用。为了支持这一假设,用高选择性SFK抑制剂eCF50629处理MDA-MB-231 TNBC细胞阻断了peak1增强的PLC-γ-1酪氨酸磷酸化。然而,对BiCAP数据集的查询没有揭示特异性SFK或PLC-γ-1与任何PEAK复合物的关联,强烈表明调节机制不涉及直接结合。
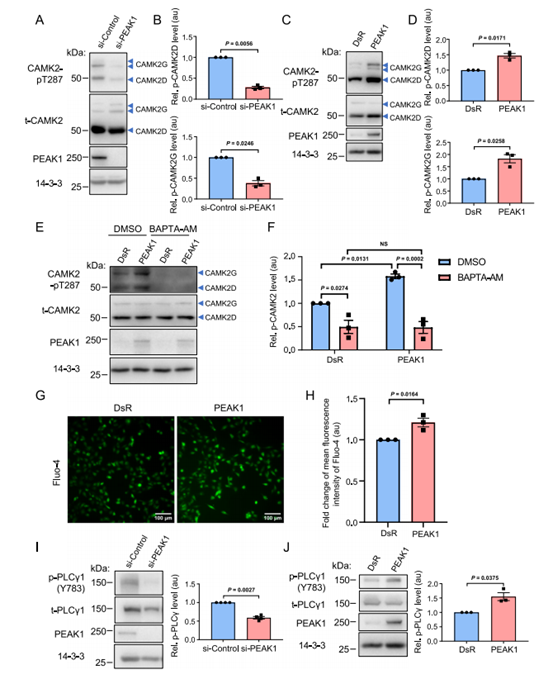
图2 PEAK1介导的CAMK2活性在三阴性乳腺癌细胞中的调节
4. PEAK1/2与CAMK2相互作用的结构要求
为了确定PEAK1和PEAK2的哪些区域介导了与CAMK2D和G的相互作用,我们利用了先前描述的PEAK1和PEAK216的缺失突变体,这些突变体删除了n端区域(ΔN-term), N1 α-螺旋(ΔN1)以及假激酶结构域和螺旋J-M的组合(ΔCterm),以及仅包含极端n端区域(1-324个氨基酸)的突变体(n项)(图3A)。在WT、ΔN1和ΔCterm PEAK1中,可以证明flag标记的PEAK1与内源性CAMK2D共免疫沉淀,但在ΔNterm PEAK1突变体中则没有(图3B)。然而,单独的Nterm区域没有与CAMK2D关联,这表明它是与这些CAMK2s相互作用所必需的,但不是充分的(图3B)。此外,由于N1和Cterm区域是PEAK1与PEAK216进行同型结合和异型相互作用所必需的,这些数据还表明,PEAK1与这些CAMK2s结合不需要二聚化。接下来,我们生成了一系列缺失突变体,以更窄地定义PEAK1上的CAMK2结合区(图3C)。与CAMK2D的关联在Δ261-324突变体中丢失(图3D)。此外,删除跨越261-300或301-324氨基酸的较小区域也会消除CAMK2D关联(图3D)。氨基酸301-324的缺失也显著降低了CAMK2G的关联(图3E)。
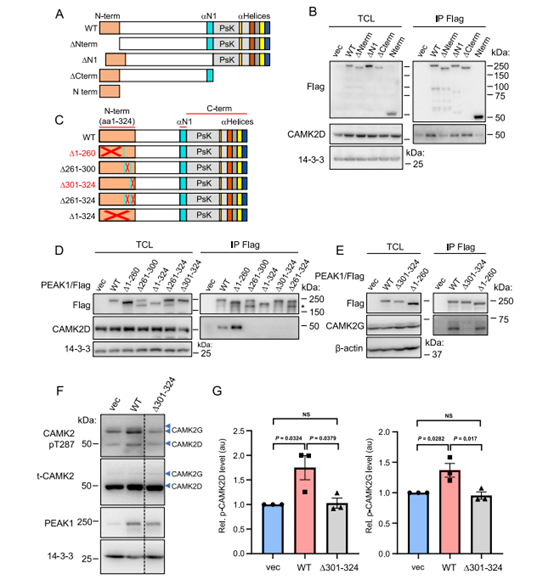
图3 绘制CAMK2在PEAK1上的结合区
尽管PEAK1和PEAK2具有相似的整体结构域结构,但n端扩展,包括CAMK2结合所需的PEAK1区域(氨基酸261-324),保守性较差,表明PEAK2的CAMK2结合机制可能不同。与这一假设相一致的是,虽然PEAK2 ΔNterm突变体显著降低了CAMK2D与PEAK2的关联,但在ΔN1和ΔCterm突变体中观察到的影响最大,它们分别显著且几乎完全消除了这种关联。由于N1和Cterm区域是PEAK2进行同型和异型结合所必需的,这些数据表明PEAK2与这些CAMK2s的相互作用需要PEAK2二聚化。为了支持这一模型,PEAK2中的三个点突变消除了二聚化,都导致与CAMK2D的关联减弱。这与PEAK1形成对比,在PEAK1中,二聚化受损的ΔN1和ΔCterm突变体保留了与CAMK2s的关联。此外,PEAK1敲低并没有破坏PEAK2与CAMK2D的关联,这表明PEAK2与CAMK2D的相互作用不是通过PEAK1介导的,这与BiCAP的数据一致(图1B)。综上所述,这些数据突出了PEAK1和PEAK2与CAMK2关联的不同结构要求,在PEAK1的情况下,来自离散n端区域的贡献更大,而在PEAK2的情况下,则需要二聚化。
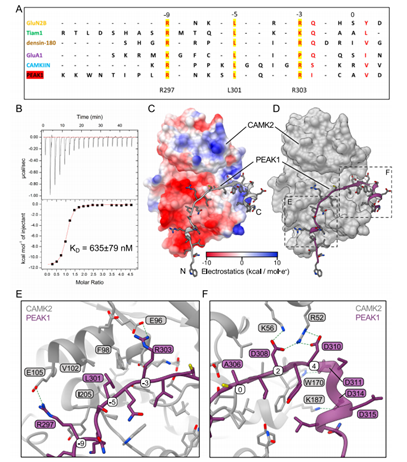
图4 PEAK1中保守CAMK2相互作用基序(CIM)的特征
5. PEAK1通过保守基序直接结合CAMK2,促进CAMK2活化和PEAK1磷酸化
由于PEAK1 Δ301-324突变体缺乏CAMK2关联(图3D,E),我们确定了该突变体对CAMK2激活的影响。有趣的是,该突变体激活CAMK2D/G的能力明显受损,这是通过T287位点的磷酸化来确定的(图3F,G),这表明peak1介导的CAMK2激活除了需要Ca2+信号外,还需要两种蛋白的结合(图2E,F)。
最近的一份报告描述了CAMK2如何通过结合伙伴R/K-X-X-h-x-R/K-X-X- s /T-h中的保守序列基序与底物结合伙伴相互作用(其中,h代表疏水残基),该基序结合在CAMK2激酶域上的单个连续位点上。它还揭示了保守的CAMK2相互作用基序(CIM)的结合与激酶结构域调控段对CAMK2的自抑制相竞争,这解释了某些底物(如GluN2B)将CAMK2锁定在活性构象中的能力。值得注意的是,Rac GDP/GTP交换因子TIAM1显示出保守基序的假底物版本(带有a而不是S/T),但仍然能够结合并促进其在其他位点上的磷酸化。为了确定PEAK1是否可能通过这种机制与CAMK2相互作用,我们在PEAK1的n端区域搜索保守基序。我们首先构建了跨不同物种的多序列比对,随后对其进行MEME分析以确定保守的基序。该分析表明,PEAK1氨基酸297-307与典型结合基序非常相似,但在0位(氨基酸306)有一个丙氨酸,与TIAM1观察到的结果一致(图4A)。
氨基酸297-307之间存在CIM,这与261-300和301-324氨基酸缺失导致的结合中断(图3D, E)以及Δ301-324突变体未能激活CAMK2(图3F,G)一致。为了确定是否有与PEAK1 CIM结合CAMK2对应的合成肽,我们采用了等温量热法。这些数据显示了一个高亲和力的相互作用,平均解离常数(KD)为635 nM(图4B),与TIAM1相互作用区域(1.1 μM)相当。使用AlphaFold对PEAK1/CAMK2相互作用进行建模预测(pLDDT 90.7,pTM 0.899), PEAK1 CIM与其他已知的相互作用因子一样,在CAMK2激酶结构域的相同表面上相互作用,PEAK1 R303(-3位置)和R297(-9)之间形成了关键的盐桥,与CAMK2中的谷氨酸残基(图4C-F)。此外,L301被容纳在CAMK2的疏水囊中(图4E)。我们还注意到PEAK1 D308、D310、D311和D314在CAMK2表面上预测形成的盐桥和静电相互作用(图4F)。值得注意的是,虽然该建模是对CAMK2A进行的,但结合位点在所有CAMK2亚型中都是保守的。
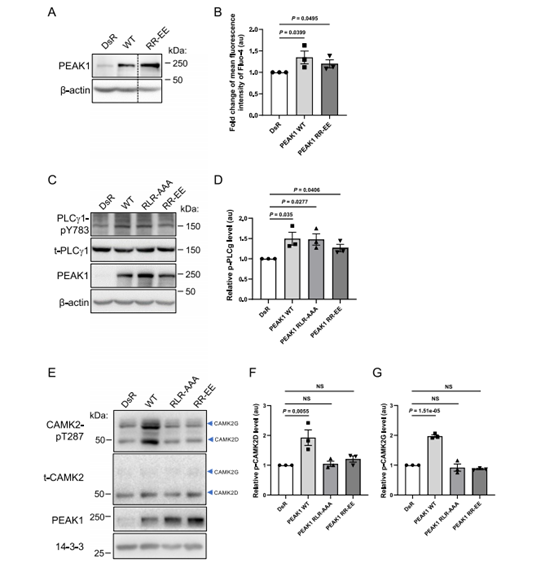
图5 PEAK1 CIM在CAMK2激活中的作用
为了测试PEAK1 CIM中关键残基对CAMK2激活的重要性,我们生成了R303(-3),L301(-5)和R297(-9)位置(称为AAA)的丙氨酸替换和R303和R297(EE)的谷氨酸替换的PEAK1突变体。EE突变体的过表达显著增强了细胞内Ca2+水平(图5A,B),两个突变体都显著增加了plc γ - 1酪氨酸磷酸化(图5C,D)。然而,两个突变体都失去了激活CAMK2的能力,这是通过T287磷酸化确定的(图5E-G)。这些数据表明,虽然PEAK1激活CAMK2的能力是钙依赖性的(图2E,F),但PEAK1增强的Ca2+不足以增加CAMK2 T287磷酸化,并且还需要与CAMK2直接关联。这些数据也支持了Alphafold模型的有效性。
CAMK2与TIAM1结合导致TIAM131的持续磷酸化。为了确定PEAK1是否是CAMK2的底物,我们使用了RA306,这是一种由赛诺菲开发的用于慢性心血管适应症的“新一代”CAMK2抑制剂,它对CAMK2D和G的选择性优于其他亚型。RA306在MDA-MB-231细胞中表现出对CAMK2的“靶向”活性(补充图7A),并显著抑制PEAK1丝氨酸/苏氨酸磷酸化(图6A,B)。此外,重组CAMK2在体外显示出对PEAK1的激酶活性,这可以用pAkt底物抗体检测到,该抗体也能识别CAMK2最小的磷酸化基元(RXXS/T)或pSer/Thr抗体(图6C)。有趣的是,当使用PEAK1 AAA和EE CIM突变体作为底物时,PEAK1的磷酸化显着降低,这表明CAMK2有效磷酸化PEAK1需要两种蛋白通过PEAK1 CIM相互作用(图6C)。
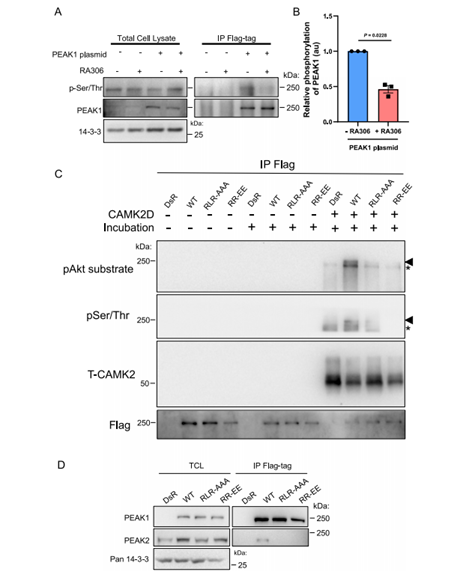
图6 CAMK2磷酸化PEAK1调控PEAK1相互作用组
6. CAMK2磷酸化PEAK1调控PEAK1相互作用组
为了表征CAMK2介导的磷酸化对PEAK1支架电位的影响,我们在体内比较了野生型(WT) PEAK1与AAA和EE CIM突变体的相互作用组。我们之所以采用这种方法,是因为它更有选择性地解决了CAMK2介导的PEAK1磷酸化的影响,而RA306治疗将对CAMK2介导的细胞磷酸化产生更普遍的抑制。适配器Crk-L和Grb2分别通过SH3和SH2结构域介导的相互作用与PEAK1结合。由于14-3-3与客户蛋白的结合典型地依赖于磷酸化,这表明CIM突变体损伤CAMK2激活导致PEAK1在14-3-3结合位点的磷酸化降低。接下来,我们分析了PEAK1与PEAK2的异型关联,这是信号输出的关键决定因素,PEAK1促进细胞迁移和致癌信号传递需要与PEAK2结合。重要的是,尽管在MDA-MB-231 TNBC细胞中可以很容易地检测到野生型PEAK1与PEAK2的异型关联,但在CIM突变体中这种异型关联明显降低(图6D)。总之,这些数据表明,CAMK2介导的PEAK1磷酸化是PEAK1介导的蛋白-蛋白相互作用和信号传导的关键调节因子。
7. PEAK1/CAMK2信号是TNBC的治疗“可操作”靶点
CAMK2和PEAK1相互调节的发现使我们质疑这些蛋白的表达如何与乳腺癌患者的生存相关。有趣的是,PEAK1和CAMK2D的高表达与较差的总生存率显著相关,而PEAK1/CAMK2G的高表达则没有明显的趋势。为了进一步了解PEAK1和CAMK2/G在乳腺癌中的联合作用,我们研究了它们与远端无转移生存(DMFS)的关系。然而,这只能通过来自某些公开可用数据集的荟萃队列(见方法)和PEAK1/CAMK2G组合来实现。接下来,我们描述了PEAK1/CAMK2信号传导对TNBC细胞生物学终点的影响。之前我们报道过,在MDA-MB-231细胞中,CRISPR介导的PEAK1 KO显著降低了低附着板中锚定依赖性增殖和肿瘤球形成。综上所述,这些数据表明PEAK1和CAMK2都有助于TNBC中的增殖信号传导。
接下来,我们探讨了PEAK1/CAMK2在细胞迁移/侵袭中的作用。CAMK2D或G的敲低均阻断了PEAK1过表达对细胞迁移和侵袭的影响(图7A、B), RA306处理阻断了PEAK1在MDA-MB-231和-468细胞中促进的迁移(图7C)和PEAK1在MDA-MB-231细胞中促进的侵袭。此外,Δ301-324 PEAK1突变体不能结合或激活CAMK2(图3D-G),也不能增强TNBC细胞的迁移和侵袭(图7D)。总的来说,这些数据表明,在调节PEAK1的细胞迁移和侵袭过程中,需要CAMK2的表达和/或激活,以及PEAK1/CAMK2的结合。
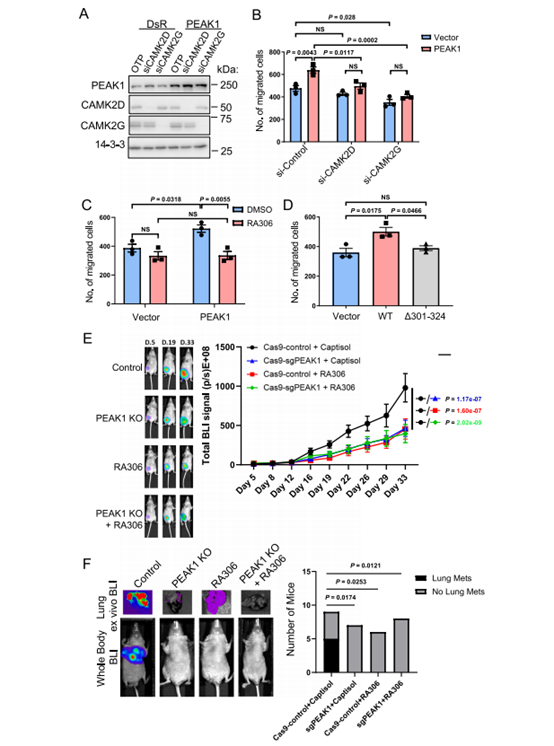
图7 CAMK2在PEAK1调控的TNBC生物学中的作用
然后,我们在TNBC肿瘤生长和转移的体内模型中评估了RA306。先前,我们证明PEAK1基因敲除可显著抑制原位MDA-MB-231 TNBC异种移植物的生长和肺转移。为了检测RA306,我们使用了该细胞系的高转移变体MDA-MB-231-HM33,因此我们可以很容易地检测原发肿瘤的转移扩散。此外,我们通过CRISPR编辑生成PEAK1缺失的MDA-MB-231-HM细胞,比较RA306与PEAK1敲除的效果以及这两种操作的联合效果。在异种移植物中可以检测到RA306的靶向活性,PEAK1消融也降低了CAMK2的激活。RA306治疗显著损害了原发肿瘤的生长(图7E)。重要的是,生长抑制程度与PEAK1基因敲除后观察到的相似,并且将RA306与基因敲除结合并没有进一步增强这种抑制程度,这强烈表明RA306治疗和PEAK1敲除针对相同的致癌信号轴(图7E)。此外,我们还允许一个单独的原发性肿瘤队列生长到相同的平均大小,切除它们,然后监测转移。引人注目的是,RA306阻断转移的程度与PEAK1敲除相似(图7F)。由于PEAK1 KO或RA306治疗影响细胞增殖、迁移和侵袭,但不影响细胞凋亡(图7),因此对原发肿瘤生长的影响可能是通过减少增殖介导的,而转移性生长的减少必须与原发肿瘤大小无关,反映了转移到肺部的能力下降和/或在继发部位的增殖减少。总的来说,这些数据确定RA306是针对TNBC和潜在的其他人类癌症的PEAK1/CAMK2轴的潜在治疗策略。
结论
本研究通过确定致癌假激酶PEAK1作为CAMK2的前馈激活剂和第二代CAMK2D/G抑制剂RA306作为阻断TNBC和其他PEAK1参与的癌症(如胰腺癌)的PEAK1/CAMK2信号传导的合适靶向药物来解决这些问题。重要的是,RA306避免了中枢神经系统的副作用,并表现出良好的口服生物利用度,突出了其肿瘤再利用的潜力。此外,相互激活的PEAK1/CAMK2复合体的鉴定表明,这两种蛋白可以作为预测性生物标志物,指导患者分层进行抗CAMK2治疗。总的来说,我们的工作确定了激酶超家族之间意想不到的相互作用,这对我们理解正常和疾病状态下的细胞信号传导以及PEAK1和CAMK2的精确靶向具有重要意义。
参考文献:
Yang, X., Ma, X., Zhao, T. et al. Activation of CAMK2 by pseudokinase PEAK1 represents a targetable pathway in triple negative breast cancer. Nat Commun 16, 1871 (2025).
实验方法:
细胞系和组织培养、细胞裂解、免疫沉淀和免疫印迹、PEAK1敲除细胞的产生、转染、BiCAP纯化、基于质谱的蛋白质组学分析、蛋白质表达和纯化、肽合成、等温滴定量热法、PEAK1(291-320)的比较建模:CAMK2、PEAK1 n端跨物种多序列比对、接近结扎试验(PLA)、钙含量测定、增殖试验、Transwell迁移/侵袭试验、体外激酶测定,肿瘤组织均质。
