






靶向RTN4的内质网膜重塑诱导焦亡以促进抗肿瘤免疫
焦亡是一种已确定的程序性细胞死亡,与内质网(ER)动力学高度相关。然而,目前尚不清楚调节触发焦亡的动态ER膜曲率变化的关键蛋白质。本研究合成了一种生物素标记的强效焦亡诱导剂α-山竹素(α-MG)化学探针。通过蛋白质微阵列分析,网织蛋白-4(RTN4/Nogo)作为ER膜曲率的关键调节因子,被确定为α-MG的靶点。作者观察到,α-MG通过募集E3连接酶UBR5化学诱导RTN4的蛋白酶体降解显著增强了癌症细胞的焦下垂表型。有趣的是,RTN4表达的下调显著促进了ER膜曲率的动态重塑,从小管过渡到片状,从而导致ER与细胞质膜的快速融合。特别是,观察到几个关键的ER标记物向焦亡细胞的“气泡”结构的易位支持了ER与质膜的融合过程。此外,α-MG诱导的RTN4敲除导致丙酮酸激酶M2(PKM2)依赖的常规半胱氨酸天冬氨酸蛋白酶-3/胃泌素E(GSDME)裂解,从而导致焦亡进展。在体内,作者观察到化学或基因RTN4敲低显著抑制癌症细胞生长,这进一步表现出抗程序性死亡-1(抗PD-1)的抗肿瘤免疫反应。在转化研究中,RTN4的高表达与肿瘤转移和患者死亡密切相关。综上所述,RTN4通过调节ER膜曲率重塑在诱导焦亡中起着重要作用,因此是抗癌免疫治疗的一个有前景的药物靶点。该研究于2025年2月发表在《Protein & Cell》,IF 13.6分。
技术路线:

主要研究结果:
1 RTN4作为焦亡功能蛋白的发现
化学遗传学是利用生物活性小分子作为化学工具发现功能蛋白的关键策略。通过基于焦亡表型的高内涵筛选,作者首次发现α-莽果素(α-MG),一种天然衍生的黄酮衍生物,是一种有效的化学探针,能够诱导显著的焦亡表型(图 1A 和 S1A)。在这里,作者选择了表现出最显著表型变化的骨肉瘤细胞进行进一步研究。作者观察到,α-MG 以浓度和时间依赖性的方式通过 Annexin V-PI 双染色(图 1B)、流式定量分析(图 1C)和乳酸脱氢酶(LDH)释放实验(图 1D)增加了 U2OS 细胞中焦亡的比例。免疫印迹分析还证实,α-MG 诱导了典型的 caspase-3 裂解 GSDME(图 1E),表明这是一种有效的化学工具,可用于研究焦亡生物学。具体来说,为了研究 GSDME 介导的焦亡是否是 α-MG 诱导的细胞死亡的主要机制,作者利用小干扰 RNA(siRNA)进行敲低实验,有效抑制 GSDME 的表达。如图 1F-H 所示,沉默 GSDME 导致 α-MG 诱导的焦亡体形成和 LDH 释放显著逆转,表明焦亡依赖于 GSDME。此外,作者还检测了不同骨肉瘤细胞(U2OS、HOS 和 143B)和正常成骨细胞系 MC3T3 以及临床骨肉瘤样本中 GSDME 的表达水平。如图 S1B 所示,这些骨肉瘤细胞中的 GSDME 表达显著高于对照组 MC3T3 细胞,表明骨肉瘤可能包含一个表现出较高 GSDME 表达水平的细胞亚群。同时,在临床骨肉瘤样本中也观察到了 GSDME 水平的存在,证实了细胞培养研究的结果(图 S1B)。此外,作者还通过免疫组化检测了来自 102 例原发性骨肉瘤患者的石蜡包埋标本中的 GSDME 表达。结果显示,与肿瘤旁组织相比,骨肉瘤中 GSDME 的表达较高。特别是,骨肉瘤组织中 GSDME 的高表达与患者 3 年内肿瘤转移和死亡呈正相关(图 S1C-E)。与此同时,骨肉瘤组织中 caspase-3 的水平在与肿瘤转移和患者 3 年内死亡率相关方面并未表现出显著差异(图 S1D 和 S1F)。
鉴于 caspase-3 和 GSDME 是细胞焦亡中的两个关键因素,作者提出 caspase-3 可能是促进骨肉瘤细胞焦亡的 GSDME 的潜在协同因子。因此,GSDME 可以作为研究骨肉瘤发病风险的评估因素,而 caspase-3 可以与之结合用于评估。总体而言,这些结果表明 α-MG 倾向于诱导高表达 GSDME 的骨肉瘤细胞发生焦亡。由于 α-MG 之前已被证实可以抑制 IDH1-R132H,作者建立了过表达 IDH1-R132H 的 U2OS 细胞,并随后通过 Annexin V-PI 双染色评估 α-MG 对细胞焦亡的影响。作者观察到,与野生型对照 U2OS 细胞相比,过表达 IDH1-R132H 的 U2OS 细胞并没有导致 α-MG 诱导的焦亡表型发生显著变化(图 S1G 和 S1H)。此外,作者还利用经典的 IDH1-R132H 抑制剂 AGI-5198 处理 U2OS 细胞,并在 α-MG 存在的情况下进行实验。作者的结果显示,AGI-5198 并没有显著影响 α-MG 在 U2OS 细胞中诱导的焦亡表型,如通过 Annexin V-PI 双染色评估的那样(图 S1I 和 S1J)。因此,作者提出 IDH1-R132H 可能不是 α-MG 诱导的 U2OS 细胞焦亡的主要贡献者。为了探索 α-MG 的生物学靶点,作者合成了生物素标记的 α-MG(生物素 - α-MG,图 S2A-F),以发现潜在的结合蛋白,使用 HuProt 蛋白组微阵列,随后与 Cy3-标记的链霉亲和素孵育(图 1I)。结果表明,被鉴定为与 α-MG 结合的蛋白质中,信号与噪声比(SNR)最高的蛋白质是网膜蛋白 - 4(RTN4)(图 1J)。众所周知,RTN4 包含三种主要亚型:RTN4A、RTN4B 和 RTN4C,它们是内质网曲率的关键调节因子。在这里,作者发现通过癌症基因组图谱(TCGA)数据库分析,RTN4B 是在各种肿瘤中表达的主要 RTN4 亚型,特别是在肉瘤中(图 S1K)。
接下来,作者发现生物素 - α-MG 主要捕获了 RTN4B 和少量的 RTN4C,通过竞争性拉下实验(图 1K)。因此,作者专注于 RTN4B 进行进一步研究。微量热泳(MST)实验表明,α-MG 和生物素 - α-MG 都能直接与 RTN4 结合(α-MG KD = 12.1 ± 1.28 nmol/L,生物素 - α-MG KD = 39.0 ± 1.09 nmol/L)(图 1L 和 S2G)。此外,分子对接分析揭示了 α-MG 通过与残基如谷氨酸(GLU)34、GLU 37 和丙氨酸 341 形成多个氢键,在 RTN4 周围形成空间键合(图 1M)。同样,生物素 - α-MG 也通过与 GLU 34、GLU 37 和 GLU 41 建立几个氢键与 RTN4 结合(图 S2H)。总之,作者确定 RTN4 是 α-MG 的直接结合蛋白,与焦亡进展高度相关。
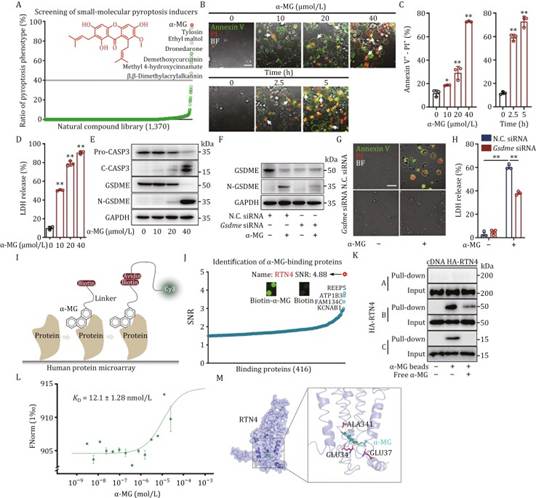
图1:发现 RTN4 作为焦亡中的功能性蛋白
2 RTN4缺乏显著促进焦亡表型
为了确定 RTN4 是否在焦亡进展中发挥作用,作者在骨肉瘤细胞系(U2OS、HOS 和 143B)中进行了 RTN4 沉默实验,并通过 Annexin V-PI 双染色观察到显著的焦亡相关“气泡”形态(图 2A 和 S3A)。其他细胞系如 HeLa 和 MGC803 也观察到了类似的表型(图 2A)。此外,流式细胞仪分析显示,RTN4 siRNA 显著提高了 Annexin V-PI 阳性焦亡细胞的比例(图 2B 和 S3B)。接下来,作者构建了一个基于 lentiviral miR30 的 Tet 诱导型(pLKO-Tet-On)短发夹 RNA(shRNA)系统,以特异性地在 U2OS 细胞中敲低 RTN4(图 S3C 和 S3D)。如图 2C 所示,多西环素(dox)显著诱导了 U2OS 细胞中的典型焦亡“气泡”形态,通过 Annexin V-PI 双染色(图 2D)。此外,dox 处理增加了 U2OS 细胞中的 LDH 释放(图 2E),表明 RTN4 缺失对焦亡进展有重要贡献。接下来,作者探索了 RTN4 敲低与各种细胞应激刺激的协同效应。作者观察到,RTN4 敲低增强了 TNF-α/环己酰亚胺(CHX)(Hu 等,2020)或顺铂(DDP)(Wang 等,2017)处理的 U2OS 细胞中的焦亡表型(图 2F)。与此同时,HeLa 和 MGC803 细胞在 RTN4 敲低后对焦亡的敏感性增加(图 S3E)。这些观察结果通过定量流式细胞仪测量(图 2G)和 LDH 释放实验(图 2H)得到了进一步证实。随后,缺氧(Yu 等,2019)和紫外线辐射(Liu 等,2021)等物理刺激也显示出在 RTN4 敲低存在的情况下对焦亡诱导的协同效应(图 2I-K 和 S3F)。有趣的是,化学和物理应激均下调了 RTN4 的表达,这代表了焦亡过程中的一种普遍生物学反应(图 2L)。此外,免疫印迹实验表明,RTN4 缺失显著激活了 caspase-3 以裂解 GSDME(图 2M 和 S3G),并随后在 RAW264.7 巨噬细胞中引发强烈的免疫反应,通过增强 TNFα、IL-6、iNOS 和 IL-1β 等炎症基因的表达(图 2N 和 S3H)。最后,作者在 TetOn-RTN4 shRNA U2OS 稳定细胞系中进行了拯救实验,以阐明介导焦亡的 RTN4 亚型。值得注意的是,作者的结果表明,在图 S3I 中,过表达 RTN4B 有效地恢复了 caspase-3 和 GSDME 的裂解,而转染 RTN4A 或 RTN4C 质粒则没有产生类似的效果。总之,这些发现强烈表明 RTN4B 在调节焦亡中起着关键作用。
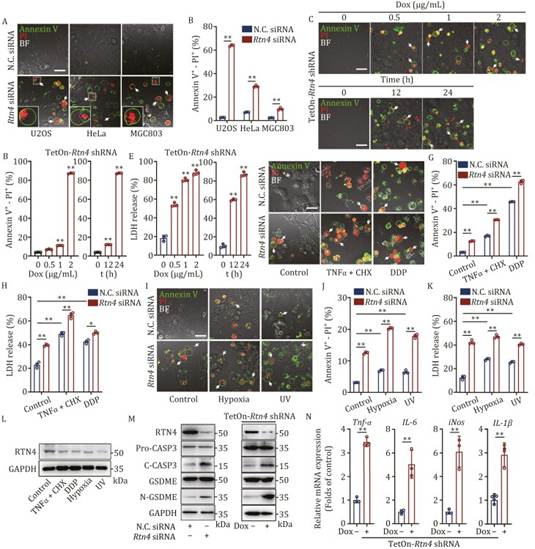
图2:RTN4 缺陷显著促进细胞焦亡表型
3 α-MG通过在泛素-蛋白酶体系统中募集E3连接酶UBR5作为RTN4降解剂
接下来,作者研究了 α-MG 对 RTN4 表达的影响。有趣的是,作者观察到 α-MG 以浓度和时间依赖性的方式显著降低了 RTN4 蛋白的表达(图 3A 和 S4A),但并未影响 mRNA 水平(图 S4B)。然后,作者使用蛋白合成抑制剂环己酰亚胺(CHX)阻断 RTN4 蛋白合成,并发现 α-MG 处理后 RTN4 表达仍然被下调,这表明 α-MG 作为一种 RTN4 化学降解剂(图 3B)。随后的 MG132 和巴非霉素 A1(BafA1)处理表明,α-MG 主要通过蛋白酶体途径而非溶酶体途径降解 RTN4(图 3C)。鉴于 RTN4 的降解主要依赖于蛋白酶体途径,作者接下来研究了 RTN4 是否经历了泛素化修饰。西方印迹分析显示,α-MG 显著促进了 RTN4 的泛素化修饰(图 3D)。特别是,这种泛素化修饰主要通过 K48 依赖的泛素链形成介导,对 K63 泛素链形成的影响较小(图 3E)。作者随后假设 α-MG 通过招募特定的 E3 泛素连接酶来促进 RTN4 的 K48 依赖性泛素化修饰。因此,作者在 MG132 处理后对 HA 标签的 RTN4 进行免疫沉淀,并通过质谱(MS)鉴定相互作用蛋白(图 3F)。然后,作者鉴定了 1290 种与 RTN4 相互作用的细胞内蛋白。在筛选 RTN4 的关键泛素连接酶后,作者最终鉴定了 6 种 E3 泛素连接酶,包括 TRIM21、UBR5、TRIM28、TRAF7、CHIP 和 TRIP12(图 3F)。与此同时,作者还鉴定了 E1 泛素激活酶 UBA1 和 E2 泛素酶 UBE2D3(图 3F)。为了进一步确定哪种 E3 泛素连接酶参与 RTN4 的泛素化,作者使用 siRNA 敲低这些 E3 泛素连接酶的表达。然后,作者用 α-MG 处理细胞,发现只有在 UBR5 敲低时,α-MG 诱导的 RTN4 降解才被逆转(图 3G)。此外,UBR5 敲低有效地阻碍了 α-MG 诱导的焦亡体形成(图 S4C)。此外,作者观察到 UBR5 过表达在 U2OS 细胞中诱导了“气泡”形态,通过 Annexin V-PI 双染色评估并经流式细胞仪分析定量(图 3H 和 3I)。因此,作者得出结论,α-MG 可能促进了 UBR5 和 RTN4 之间的类似分子胶的相互作用,随后通过 K48 依赖途径增强了下游蛋白酶体对 RTN4 的降解。
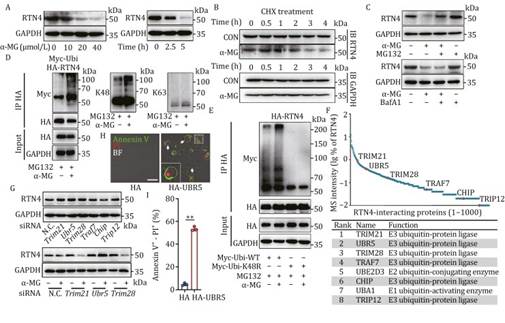
图3:α-MG 通过在泛素-蛋白酶体系统中募集 E3 连接酶 UBR5 作为 RTN4 降解剂
4 RTN4敲除通过管状到片状变化重塑ER膜曲率
由于 RTN4 是调节内质网膜曲率的关键蛋白,因此 RTN4 缺失可能会对内质网的形态特征产生影响。在这里,共聚焦荧光分析显示,通过 siRNA 敲低 RTN4 诱导 U2OS 细胞中内质网形态从网格状管状结构转变为扁平片状结构,这些细胞转染了内质网标记物 Sec61β-EGFP 和 Lys-Asp-Glu-Leu(KDEL)-pDsRed2(图 4A)。此外,作者观察到化学诱导的 RTN4 降解通过 α-MG 也显著触发了内质网管状到片状的转变(图 4B)。此外,作者通过透射电子显微镜(TEM)分析证实,在 RTN4 siRNA 和 α-MG 处理的 U2OS 细胞中,长度超过 10μm 的片状内质网比例显著增加(图 4C 和 4D)。由于 atlastin-1 是管状内质网形成的关键介质,作者还进行了定量实时 PCR(qPCR)分析,发现 RTN4 沉默的 U2OS 细胞以及 HeLa 和 MGC803 中 atlastin-1 的 mRNA 表达显著下调(图 4E),表明内质网管状到片状的转变与细胞内 RTN4 的表达高度相关。最近的证据表明,RTN4 形态失衡可能会诱导内质网应激(Kuang 等,2006;Oertle 等,2003)。然后,作者通过 Annexin V-PI 双染色观察到,内质网应激抑制剂牛磺胆酸(TUDCA)或 4-苯基丁酸(4-PBA)逆转了 RTN4 敲低诱导的类似“气泡”的焦亡表型(图 4F)。与此同时,内质网应激诱导剂硫代巴比妥酸(TG)或鹅去氧胆酸(TM)显著增强了 U2OS 细胞中的“气泡”形态(图 4F)。这些发现也通过流式细胞仪(图 4G)和 LDH 释放实验(图 4H)得到了证实,表明内质网应激在 RTN4 缺失诱导的焦亡中具有关键的桥梁功能。
接下来,为了深入了解 RTN4 与内质网形态转变之间的关系,作者进行了共免疫沉淀(co-IP)实验,以捕获细胞中的 RTN4 结合蛋白。然后,96 种蛋白显著富集,并被分为五类,包括细胞骨架动态、膜流动性、转录因子、内质网应激和其他(图 4I)。在这些 RTN4 结合蛋白中,丙酮酸激酶 M2(PKM2)特别引起了作者的兴趣。由于 PKM2 之前已被报道与通过调节内质网应激调节焦亡进程高度相关,并且因此被认为是进一步研究的关键候选蛋白。为了测试 RTN4 与 PKM2 的相互作用,作者进行了 co-IP 实验,并发现 RTN4 直接与 PKM2 结合(图 4J)。与此同时,α-MG 的处理导致 PKM2 表达下调,最终导致 GSDME 的裂解(图 4K)。此外,通过微量热泳(MST)分析定量表征 RTN4 与 PKM2 之间的亲和力,解离常数(KD)为 151.12 ± 16.84 nmol/L(图 4L),表明 RTN4 与 PKM2 之间存在强烈的相互作用。此外,PKM2 敲低在 U2OS 细胞中引发了“气泡”形态,通过 Annexin V-PI 双染色(图 4M 和 4N)。与此同时,PKM2 敲低促进了 LDH 释放,最终通过诱导 caspase-3/GSDME 激活触发焦亡(图 4O-Q)。接下来,为了研究 PKM2 对内质网膜曲率的影响,作者在表达内质网标记 Sec61βEGFP 的 U2OS 细胞中进行了共聚焦荧光分析。PKM2 敲低未能诱导内质网管状到片状的转变(图 S5A)。此外,作者观察到 UBR5 过表达诱导内质网形态从网格状管状结构转变为扁平片状结构(图 S5B)。与此同时,通过 siRNA 敲低 UBR5 对内质网膜曲率的改变没有明显影响(图 S5C)。因此,这些发现表明 UBR5 而不是 PKM2 是调节内质网膜曲率重塑的上游信号通路。最后,作者进行了实验,深入探讨内质网膜曲率重塑与 caspase-3/GSDME 信号之间的相关性。作者的结果表明,通过使用 Z-DEVDFMK 抑制 caspase-3 或通过 siRNA 耗尽 GSDME,并没有对内质网膜曲率产生任何显著影响,例如“管状到片状”的转变(图 S5D 和 S5E)。这些发现表明,caspase-3/GSDME 信号可能位于内质网膜曲率重塑的下游。总之,作者的发现表明,RTN4 缺失诱导的内质网膜曲率变化通过内质网应激信号通路导致焦亡。
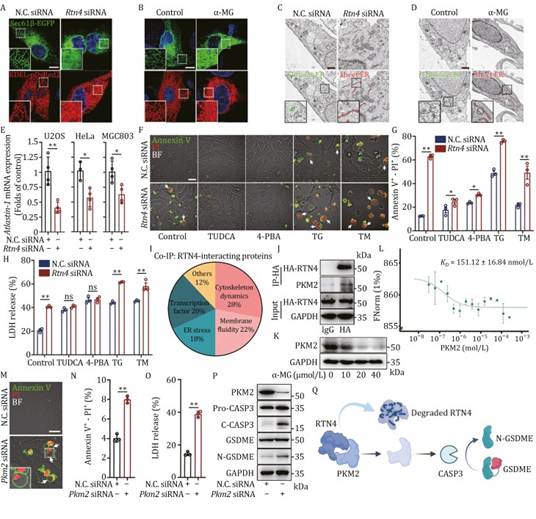
图4:RTN4 敲低通过肾小管到片层的变化重塑 ER 膜曲率
5 在焦亡过程中,膜曲率的变化驱动内质网融合到“气泡”结构
“气泡”结构的形成是焦亡中的一个重要生物学事件(Chen 等,2016)。为了探索内质网在 RTN4 缺失诱导的类似“气泡”的表型中的作用,作者应用了一系列细胞器荧光探针(内质网追踪剂、线粒体追踪剂、溶酶体追踪剂、高尔基体追踪剂和细胞膜追踪剂 DiI)。有趣的是,作者发现 RTN4 敲低诱导的“气泡”结构明显被内质网追踪剂标记,但未被线粒体追踪剂或溶酶体追踪剂标记;而这些“气泡”结构被高尔基体追踪剂和 DiI 轻微标记(图 5A)。相应地,作者还通过透射电子显微镜(TEM)成像证实了 α-MG 处理后“气泡”结构中内质网的存在(图 5B)。接下来,为了确认内质网与“气泡”结构之间的直接联系,作者在 U2OS 细胞中过表达内质网标记物(Sec61β、Calnexin、KDEL)。活细胞成像分析显示,通过 siRNA 敲低 RTN4 后,Sec61β-EGFP 的绿色荧光出现在“气泡”结构中(图 5C)。类似的结果也在多西环素诱导的 RTN4 敲低模型中得到证实(图 5D)。此外,Calnexin-EGFP 标记的内质网膜和 pDsRed2-KDEL 标记的内质网腔在 RTN4 敲低细胞的“气泡”结构中也高度表达(图 5E 和 5F)。RTN4 降解剂 α-MG 在荧光共聚焦中模拟了遗传性 RTN4 敲低,将这些内质网标记物转移到“气泡”结构中(图 S6B 和 S6C)。此外,RTN4 沉默显著促进了 Sec61β-EGFP 标记的内质网膜与 GSDME-N 孔在“气泡”结构周围的共定位(图 S6D)。与此同时,RTN4 降解剂 α-MG 展示了诱导 Sec61β 标记的内质网膜转移的类似效果(图 S6E)。此外,免疫印迹实验表明,内质网蛋白(Sec61β、Calnexin 和 KDEL)在响应 RTN4 敲低时从细胞质释放到培养上清液中(图 5G),进一步支持了作者的假设,即内质网可能在焦亡过程中通过促进其膜与“气泡”结构融合来发挥作用。值得注意的是,脂质体结合实验表明,纯化的片状内质网蛋白而非管状内质网蛋白对类似细胞内膜的脂质体具有结合偏好(图 5H、5I 和 S6F)。此外,内质网膜蛋白,包括 Sec61β 和 Calnexin,主要存在于由片状内质网成分构成的脂质体结合沉淀物中(图 5J)。总之,这些数据表明内质网在焦亡进程中高度参与,通过促进其膜与“气泡”结构融合来发挥作用。
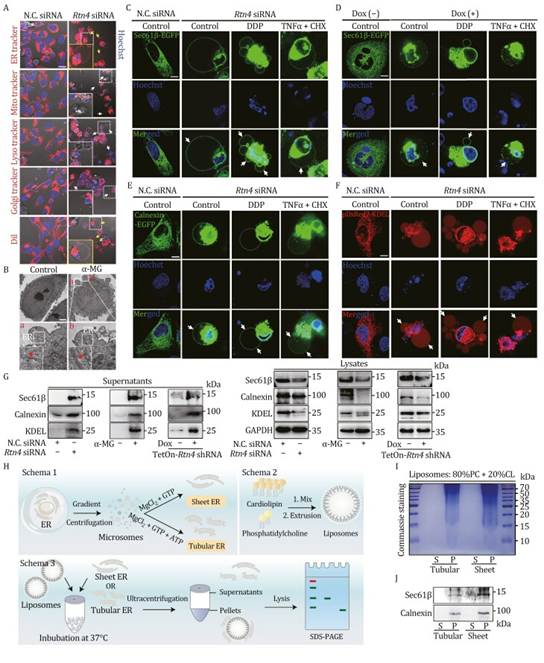
图5:RTN4 敲低促进 ER 膜融合到“气泡”结构
6质膜紊乱协同促进RTN4缺乏诱导的焦亡
由于内质网膜可能通过内质网 - 细胞膜融合参与构建“气泡”结构,作者推测细胞膜稳态可能影响焦亡进程。为此,作者使用两性表面活性剂 Triton X-100 轻度处理细胞,部分去除 Triton X-100 可溶解的细胞膜成分(Adachi 等,2007)。如图 6A 所示,Triton X-100 单独使用并未在 U2OS 细胞中引发明显的焦亡;然而,Triton X-100 显著增加了多西环素诱导的“气泡”结构比例,通过 Annexin V-PI 双染色和流式分析(图 6B)。此外,Triton X-100 也显著促进了多西环素诱导的 U2OS 细胞中的 LDH 释放(图 6C)。同样,Triton X-100 加速了 RTN4 降解剂 α-MG 诱导的 U2OS 细胞中的“气泡”结构形成(图 6D-F)。此外,GSDME-N 过表达增强了 α-MG 诱导的焦亡中的“气泡”结构形成(图 6G 和 6H)。总之,这些结果表明细胞膜稳态在促进 RTN4 敲低诱导的焦亡中的“气泡”结构形成中发挥着重要作用。
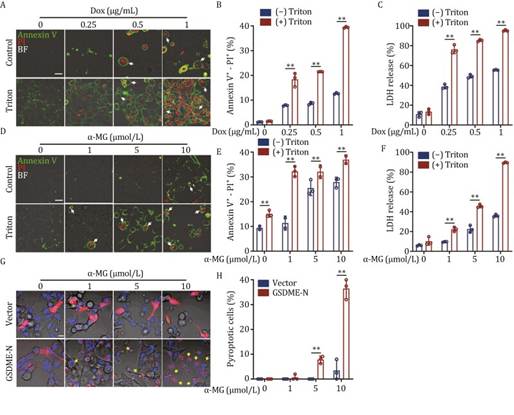
图6:质膜干扰协同促进 RTN4 缺陷诱导的焦亡
7 RTN4降解剂在抗癌治疗中的转化研究
为了评估针对 RTN4 的抗癌潜力,作者建立了 3D 肿瘤球体模型,并用 Calcein-AM 和 EthD-1 进行染色(Kamoshima 等,2011)。如图 7A 所示,多西环素在抑制 3D 肿瘤模型生长方面表现出浓度依赖性抗肿瘤活性。RTN4 降解剂 α-MG 也显示出类似的结果(图 7A)。接下来,作者利用表达 RTN4 shRNA 的 K7M2 细胞建立了骨肉瘤异种移植小鼠模型,该模型由多西环素激活。作者发现,通过肿瘤内注射多西环素,可以通过下调 RTN4 表达有效抑制肿瘤形成(图 7B、7C、S7A 和 S7B)。接下来,为了探索 RTN4 降解用于抗癌治疗的临床转化可行性,作者测试了 RTN4 降解剂 α-MG 对携带 K7M2 肿瘤的小鼠的口服给药的治疗效果。作者观察到,α-MG(50 或 100 mg/kg)显著减少了 K7M2 肿瘤的大小(图 S7C 和 S7D)和重量(图 S7E),而没有体重减轻(图 S7F)。此外,免疫组化(IHC)分析显示,α-MG 在肿瘤组织中显著上调了 T 细胞标记物 CD8、NK 细胞标记物 CD56 以及巨噬细胞标记物 F4/80 的水平(图 S7G),表明激活了明显的抗肿瘤免疫反应。
与此同时,作者发现程序性细胞死亡 1 配体 1(PD-L1)的表达在 α-MG 处理后显著上调(图 S7G),暗示了 α-MG 与免疫检查点疗法的协同可能性。然后,作者设计了一个联合方案,将 α-MG 与抗 PD-1 抗体联合使用(图 7D)。作者的结果显示,α-MG 给药与抗 PD-1 治疗联合使用在减少肿瘤体积和重量方面显示出显著的协同作用(图 7E、7F 和 S7H),而没有对体重、器官重量和血清生化指标产生明显的不良影响(图 S7I-L)。此外,作者通过免疫组化方法检测了来自 101 例原发性骨肉瘤患者的石蜡包埋标本中的 RTN4 表达(图 7G、7H 和 S8)。此外,作者评估了骨肉瘤组织和正常骨组织的免疫组化特征,结果显示骨肉瘤组织中 RTN4 的表达相对较高(图 S7M)。因此,这些结果表明 RTN4 可能是抗癌转化医学研究中的一个可用生物标志物或治疗靶点。鉴于 RTN4 在焦亡以及骨肉瘤进展中的功能重要性,抑制 RTN4 代表了一种潜在的抗癌策略。为此,作者利用包含 10845 种化合物的 DrugBank 数据库进行了高通量虚拟筛选。每个化合物都被对接到由 I-TASSER 生成的 RTN4 结构的预测结合口袋中。随后,计算了 Glide XP GScore 值(kcal/mol),这些值代表了这些化合物与 RTN4 结合的亲和力。从这次筛选中,作者选择了排名前 20 的化合物(图 7I)进行进一步的实验验证。通过 MST 实验,作者证实了这些化合物中的大多数对 RTN4 具有很强的结合亲和力(图 S7N)。此外,通过在 U2OS 细胞中进行焦亡特征分析(明场显微镜和 Annexin V-PI 双染色)表明,米托蒽醌、克唑替尼和伊达比星显著诱导了焦亡(图 S7O、S7P 和 7J)。总之,作者证明了靶向 RTN4 诱导焦亡的潜力,为开发针对 RTN4 的治疗药物提供了先导化合物。
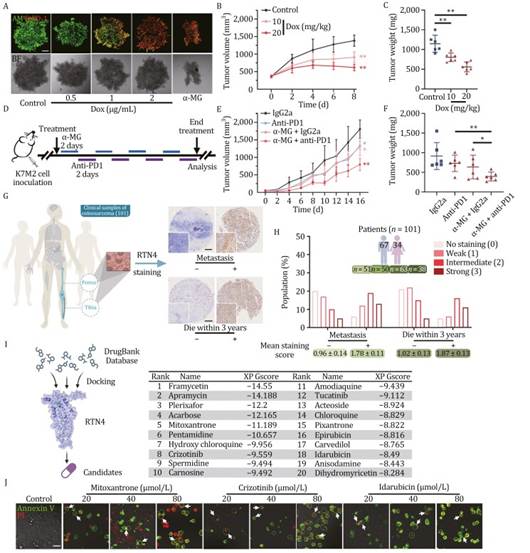
图7:RTN4 降解剂用于抗癌治疗的转化研究
结论:
总之,作者表明 RTN4 通过动态重塑 ER 膜曲率高度参与驱动焦亡。这些发现可能为作者理解焦亡过程中的基本 ER 生物学提供一个有吸引力的方向,并强调 RTN4 作为有价值的抗癌靶点的重要性。
实验方法:
免疫荧光,RT-qPCR,Western blot,免疫组化
参考文献:
Zhao MM, Ren TT, Wang JK, Yao L, Liu TT, Zhang JC, Liu Y, Yuan L, Liu D, Xu JH, Tu PF, Tang XD, Zeng KW. Endoplasmic reticulum membrane remodeling by targeting reticulon-4 induces pyroptosis to facilitate antitumor immune. Protein Cell. 2025 Feb 1;16(2):121-135. doi: 10.1093/procel/pwae049. PMID: 39252612; PMCID: PMC11786723.