






ASCT2的棕榈酰化受JNK1–ZDHHC14轴调控,协调谷氨酰胺代谢并驱动非小细胞肺癌进展
S-棕榈酰化是一种可逆的翻译后修饰,可调控蛋白稳定性和细胞功能,但其在谷氨酰胺代谢中的作用仍不清楚。本研究发现,ZDHHC14是催化ASCT2在保守的Cys39和Cys48位点发生棕榈酰化的关键棕榈酰转移酶,从而促进这一谷氨酰胺转运蛋白的溶酶体降解;而ABHD17B则作为去棕榈酰化酶发挥作用,稳定ASCT2蛋白。机制上,谷氨酰胺缺乏可激活JNK1,后者直接在Thr440位点磷酸化ZDHHC14,触发其降解,进而增强ASCT2的稳定性。重要的是,JNK抑制剂与ASCT2抑制剂联合应用在体内可协同抑制谷氨酰胺代谢和肿瘤生长。这些结果揭示了一条连接JNK介导的ASCT2棕榈酰化与谷氨酰胺代谢的“磷酸化-棕榈酰化”调控轴,为非小细胞肺癌提供了潜在的治疗策略。该研究于2026年2月发表在《Cell Discovery》,IF 12.5分。
技术路线:
主要研究结果:
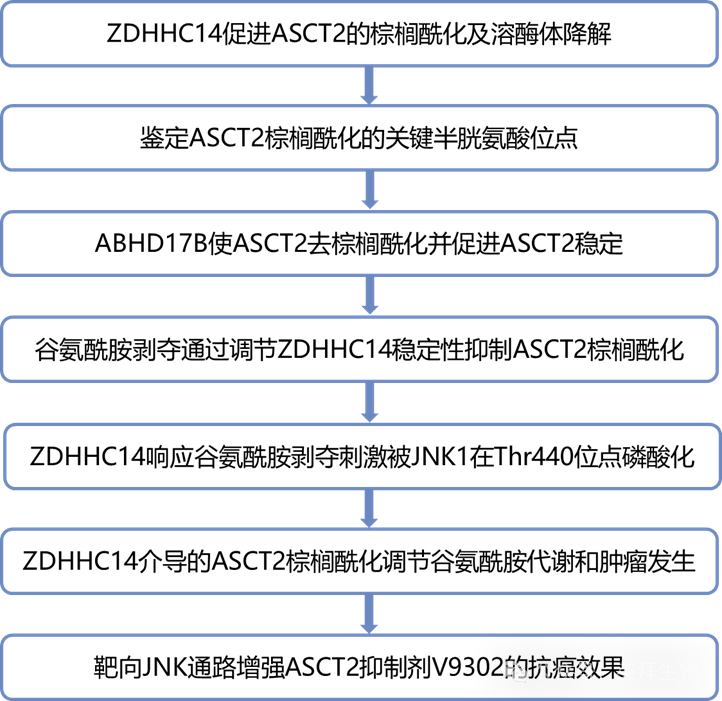
1 ZDHHC14促进ASCT2的棕榈酰化及溶酶体降解
蛋白质棕榈酰化被认为在各种代谢过程中起着重要作用。为了探究棕�酰化调控谷氨酰胺代谢的多种调节机制,研究者用 2-溴棕榈酸盐(一种通用的棕榈酰化抑制剂)处理细胞,并进行谷氨酰胺摄取实验和谷氨酸生成实验,结果显示 2-溴棕榈酸盐以剂量依赖性方式显著增强了谷氨酰胺摄取和谷氨酸生成(图 1a)。顺着这一线索,研究者对非小细胞肺癌细胞系进行了棕榈酰化蛋白质组学分析,以分析谷氨酰胺代谢调节因子的棕榈酰化情况,结果显示 ASCT2 可能被棕榈酰化(图 1b)。为了验证研究者的假设,研究者首先检测了用棕榈酸钠处理的 NCI-H1299 和 NCI-H1975 细胞中 ASCT2 的蛋白水平,发现 ASCT2 蛋白水平(而非 mRNA 水平)以时间依赖性方式显著降低(补充图 S1a)。相反,通过 2-溴棕榈酸盐抑制棕榈酰化则以剂量依赖性方式增加了 ASCT2 的蛋白表达,但不影响其 mRNA 表达。
为了探究哪种棕榈酰基转移酶负责 ASCT2 的棕榈酰化,研究者进行了免疫沉淀结合质谱分析,以鉴定潜在的 ASCT2 相互作用棕榈酰基转移酶。研究者发现了二十多种棕榈酰基转移酶候选蛋白,其中 ZDHHC14 和 ZDHHC9 被鉴定为 ASCT2 的强相互作用候选蛋白(图 1c;补充图 S1b)。通过免疫共沉淀实验,研究者证实了 ASCT2 与 ZDHHC14 和 ZDHHC9 的相互作用(补充图 S1c)。为了检测这些酶是否调节 ASCT2 的棕榈酰化,研究者进行了炔基-叠氮基点击化学实验。结果表明,在测试的八种候选棕榈酰基转移酶中,只有 ZDHHC14 显著增加了 ASCT2 的棕榈酰化水平(图 1d;补充图 S1d)。重要的是,外源性 ZDHHC14 表达显著上调了 ASCT2 棕榈酰化,而催化失活的 ZDHHC14 突变体(ZDHHC14C195S)则没有这种效果(图 1e)。此外,ZDHHC14 的过表达显著降低了 ASCT2 蛋白水平,但不影响其 mRNA 水平,而这种效应在 ZDHHC14C195S 突变体表达细胞中并未观察到(图 1f;补充图 S1e)。一致地,ZDHHC14 敲低增加了 ASCT2 蛋白水平并延长了其半衰期(图 1g-i;补充图 S1f, g)。此外,用溶酶体抑制剂巴弗洛霉素 A1 处理可阻断 ZDHHC14 介导的 ASCT2 降解,而蛋白酶体抑制剂 MG132 处理则没有这种效果(图 1j;补充图 S1h)。免疫荧光分析进一步表明,ZDHHC14 显著增强了 ASCT2 与溶酶体相关膜蛋白 1 阳性溶酶体的共定位(图 1k)。总的来说,这些结果确立了 ZDHHC14 是 ASCT2 特异性的棕榈酰基转移酶,可促进其溶酶体依赖性降解。
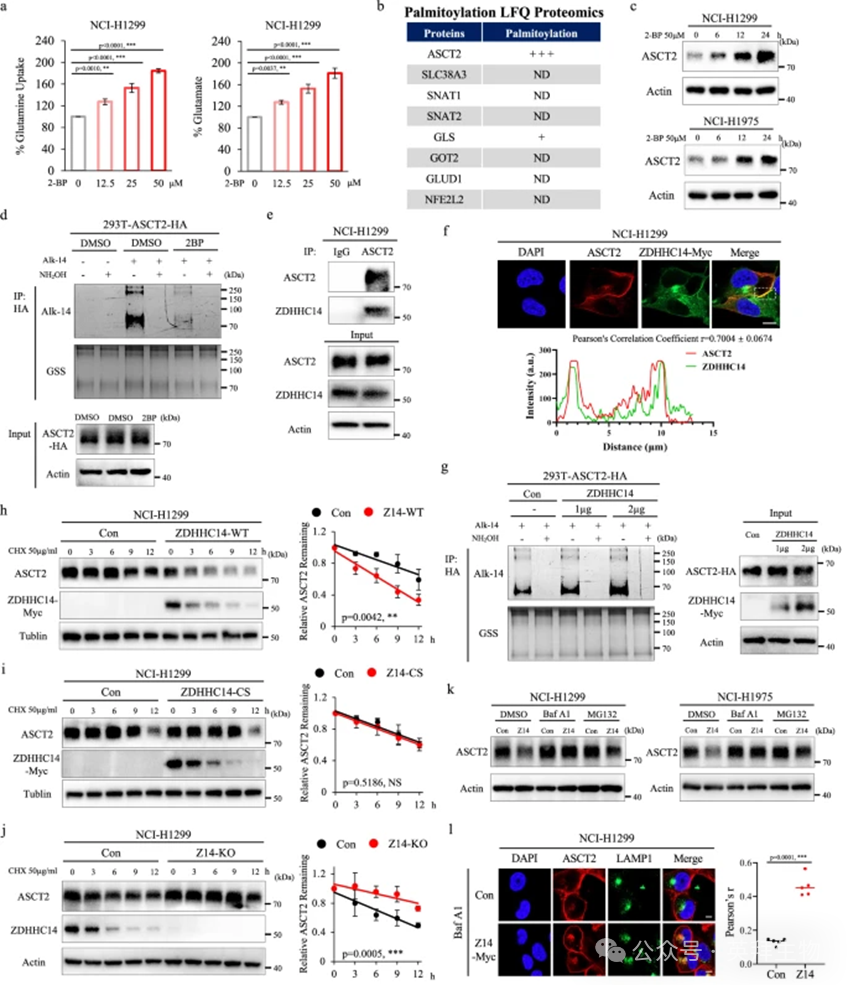
图1:ZDHHC14促进ASCT2棕榈酰化和溶酶体降解
2 鉴定ASCT2棕榈酰化的关键半胱氨酸位点
为了深入了解 ASCT2 棕榈酰化的分子机制,研究者首先分析了 ASCT2 蛋白的拓扑结构,它包含八个预测的跨膜结构域和八个可能发生棕榈酰化的胞内半胱氨酸残基(图 2a)。通过液相色谱-串联质谱分析,研究者明确鉴定了 ASCT2 中 Cys39 残基的棕榈酰化(图 2b)。为了明确鉴定 ASCT2 的棕榈酰化位点,研究者系统地将 ASCT2 蛋白中的每个半胱氨酸残基突变为丝氨酸,并通过炔基-叠氮基点击化学实验检测其棕榈酰化水平,结果显示 ASCT2C39S 和 ASCT2C48S 突变体均显著降低了 ASCT2 的棕榈酰化水平,而其他位点的单点突变(C110S, C308S, C309S, C363S, C395S, C467S)影响较小(图 2c)。同时,表达 ASCT2C39S/C48S 突变体完全消除了 ASCT2 棕榈酰化的信号,表明 Cys39 和 Cys48 位点对于 ASCT2 的棕榈酰化至关重要(图 2d)。此外,外源性 ZDHHC14 的表达显著促进了野生型 ASCT2 表达细胞中的棕榈酰化水平,但对 ASCT2C39S/C48S 突变体表达细胞无此作用,而 ZDHHC9(对葡萄糖供应至关重要,与 ASCT2 在补充图 S1c 中有强相互作用)不影响棕榈酰化水平,这表明 ZDHHC14 特异性介导 ASCT2 在 Cys39 和 Cys48 位点的棕榈酰化(图 2e;补充图 S53b)。
值得注意的是,ASCT2 的 Cys39 和 Cys48 棕榈酰化位点位于其 N 端,并且在不同物种中高度进化保守(图 2f)。为了分析 Cys39 和 Cys48 位点对 ASCT2 降解的影响,研究者用蛋白质合成抑制剂Cycloheximide处理表达野生型 ASCT2 或 ASCT2C39S、ASCT2C48S 及 ASCT2C39S/C48S 突变体的细胞,发现与野生型 ASCT2 相比,ASCT2 棕榈酰化突变体细胞中 ASCT2 的半衰期显著延长,这与 2-溴棕榈酸盐处理的效果相似(图 2g, h;补充图 S53c)。已知 ASCT2 蛋白的稳定性与其泛素化相关,而在 MG132 或巴弗洛霉素 A1 处理下,过表达 ASCT2 及其棕榈酰化突变体对泛素化水平影响很小(补充图 S53d, e)。这些发现与最近一份报告的结果一致,该报告显示一种神经元特异性蛋白 NcDN 以 N 端棕榈酰化依赖的方式定位于 Rab5 阳性早期内体。顺着这一线索,研究者通过免疫荧光分析检测了棕榈酰化对 ASCT2 定位的影响,结果显示 ASCT2C39S/C48S 突变体显著降低了 NCI-H1975 细胞中与 LAMP1 阳性溶酶体簇的共定位(图 2i)。因此,ASCT2 的 Cys39 和 Cys48 位点棕榈酰化主要参与其溶酶体介导的蛋白质降解。
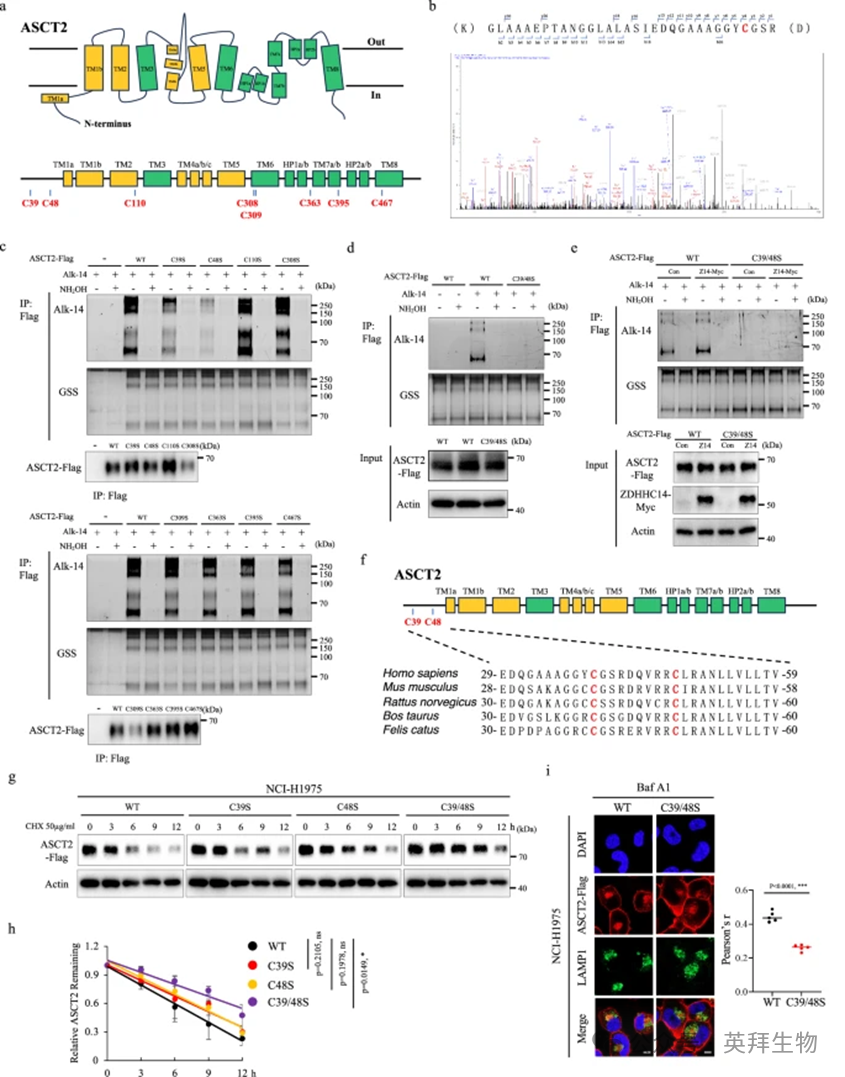
图2:ASCT2棕榈酰化的Cys39和Cys48位点主要参与溶酶体介导的蛋白质降解
3 ABHD17B使ASCT2去棕榈酰化并促进ASCT2稳定
为了鉴定 ASCT2 潜在的去棕榈酰化酶,研究者过表达了一系列已知的去棕榈酰化酶,如 ABHD17A、ABHD17B、ABHD17C、PPT1、PPT2、APT1、APT2 和 ABHD10,并通过免疫共沉淀实验进行检测,结果显示只有 ABHD17B 与 ASCT2 有强相互作用(图 3a)。在炔基-叠氮基点击化学实验中,外源性 ABHD17B 表达显著降低了 ASCT2 的棕榈酰化水平(图 3b)。为了研究 ABHD17B 是否调节 ASCT2 蛋白稳定性,研究者在 NCI-H1299 和 NCI-H1975 细胞中构建了稳定敲低 ABHD17B 表达或过表达 ABHD17B 的转染株。免疫印迹数据显示,敲低 ABHD17B 表达导致 ASCT2 蛋白水平显著降低,而外源性 ABHD17B 表达则导致其显著增加(图 3c;补充图 S54a)。为进一步了解 ABHD17B 作为去棕榈酰化酶对 ASCT2 的作用,研究者用 ABD957(一种有效的、选择性的 ABHD17A/B/C 共价抑制剂)处理,检测 ASCT2 的累积情况,结果显示 ABD957 处理导致 ASCT2 蛋白水平呈时间依赖性下降(图 3d)。此外,Cycloheximide实验证实,外源性 ABHD17B 表达抑制了 ASCT2 的降解,从而充分提高了其蛋白积累(图 3e;补充图 S54b)。此外,溶酶体抑制剂巴弗洛霉素 A1 处理,而非蛋白酶体抑制剂 MG132 处理,阻断了敲低 ABHD17B 表达细胞中 ASCT2 的降解(图 3f)。进一步的泛素化实验显示,ABHD17B 介导的 ASCT2 稳定性是通过不依赖于泛素化的途径实现的(补充图 S54c, d)。免疫荧光分析也显示,ABHD17B 表达降低了 ASCT2 与溶酶体的共定位(图 3g)。值得注意的是,研究者意外地发现 ABHD17B 和 ASCT2 并未在质膜上共定位(图 3h)。
为了深入了解 ASCT2 棕榈酰化变化的潜在机制,研究者对表达野生型 ASCT2 或 ASCT2C39S/C48S 突变体的 NCI-H1299 细胞进行了免疫沉淀-质谱分析。已有报道称,ASCT2 回收到细胞表面的过程依赖于逆转录复合物,这是一种内体回收的主要调控者。研究者首先通过免疫沉淀-质谱分析比较了 ASCT2C39S/C48S 突变体与野生型 ASCT2 的结合伙伴,发现与 ASCT2C39S/C48S 突变体结合增强的前八个候选蛋白中,有六个是依赖于逆转录复合物的内体回收组分(图 3i)。TRIM27 作为一种 RING 型 E3 泛素连接酶,通过与逆转录复合物的相互作用定位于内体,并且是 WASH 调节复合物(一种已知的逆转录复合物介导运输的调节因子)启动内体 F-肌动蛋白成核所必需的。此外,TRIM27 在非小细胞肺癌中显著升高,并与不良生存率密切相关(补充图 S54e, f)。鉴于 TRIM27 与逆转录复合物介导的运输及非小细胞肺癌的紧密关联,研究者选择它作为示例来表征 ASCT2 棕榈酰化的调节机制。为了理解 ASCT2 棕榈酰化与 TRIM27 的关联,研究者通过免疫共沉淀实验确定了野生型 ASCT2 或 ASCT2C39S/C48S 突变体对二者相互作用的影响,结果显示,表达 ASCT2C39S/C48S 突变体后,ASCT2 与 TRIM27 之间的相互作用增强(图 3j)。为了证实研究者的发现,研究者在 ABHD17B 和 TRIM27 敲低共表达的细胞中检测了 ASCT2 蛋白水平,发现敲低 TRIM27 表达显著降低了 ABHD17B 介导的 ASCT2 积累,但对其泛素化水平影响很小(图 3k;补充图 S54g)。与这些结果一致,ABHD17B 介导的 ASCT2 去棕榈酰化增强了 ASCT2 依赖于逆转录复合物的内体回收。
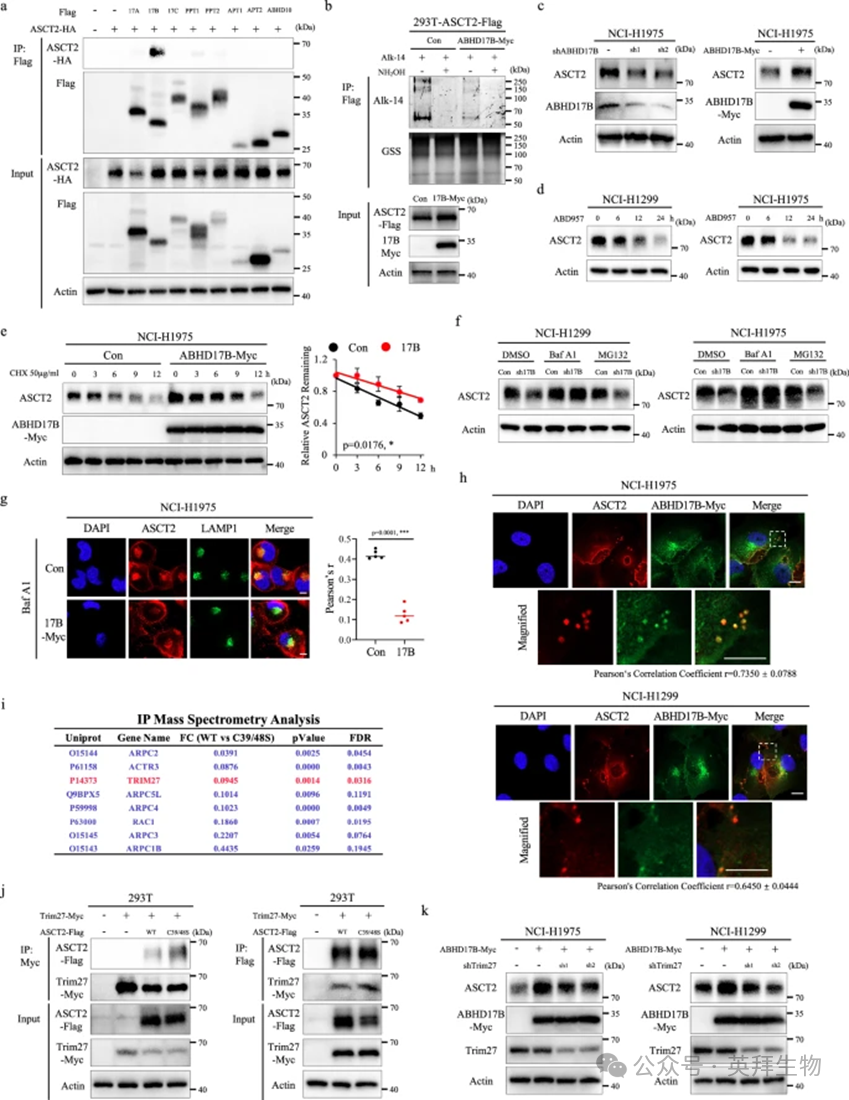
图3:ABHD17B脱棕榈酰化ASCT2并促进ASCT2稳定
4 谷氨酰胺剥夺通过调节ZDHHC14稳定性抑制ASCT2棕榈酰化
先前一项研究表明,在谷氨酰胺剥夺条件下,ASCT2 的表达会上调;然而,在这种应激条件下,棕榈酰化对 ASCT2 稳定性的影响尚未阐明。为了探究可能调节 ASCT2 棕榈酰化的应激或生理条件,研究者进行了免疫印迹分析,发现在 NCI-H1299 和 NCI-H1975 细胞中,谷氨酰胺剥夺条件下 ASCT2 的水平显著增加(图 4a)。顺着这一线索,研究者通过炔基-叠氮基点击化学实验检测了谷氨酰胺剥夺条件下 ASCT2 棕榈酰化的信号。正如预期,谷氨酰胺剥夺条件显著降低了 ASCT2 棕榈酰化的信号(图 4b;补充图 S55a)。研究者进一步观察到,在 ASCT2C39S/C48S 突变体表达细胞中,ASCT2 没有显著差异(图 4c)。为了明确证明谷氨酰胺剥夺在 ASCT2 棕榈酰化中的作用和机制,研究者随后评估了其对棕榈酰基转移酶 ZDHHC14 或去棕榈酰化酶 ABHD17B 的影响。有趣的是,在谷氨酰胺剥夺条件下,ZDHHC14 的蛋白水平降至最低,而 ASCT2 与 ABHD17B 之间的相互作用也减少,进一步表明只有棕榈酰化的 ASCT2 才能与 ABHD17B 结合(图 4a;补充图 S5b)。一致地,用Cycloheximide处理的培养细胞进一步证实,在 NCI-H1299 和 NCI-H1975 细胞中,谷氨酰胺剥夺以时间依赖性方式促进了 ZDHHC14 的降解(图 4d;补充图 S5c)。这些发现表明,谷氨酰胺剥夺通过降低 ZDHHC14 的稳定性来抑制 ASCT2 的棕榈酰化。
先前的研究已经阐明,谷氨酰胺剥夺引起的营养应激会导致 p38-MAPK、Akt-mTOR、ERK 和 JNK 通路的激活。为了解谷氨酰胺剥夺介导的 ZDHHC14 稳定性背后的分子机制,研究者在谷氨酰胺剥夺条件下,结合使用这些信号通路抑制剂,检测了 ZDHHC14 和 ASCT2 的蛋白水平,结果显示,在 NCI-H1299 和 NCI-H1975 细胞中,只有 JNK 抑制剂处理不仅提高了 ZDHHC14 的水平,还降低了 ASCT2 的水平(图 4e;补充图 S5d)。研究者注意到 p38 抑制剂也导致 ZDHHC14 积累,这是因为 p38 抑制剂处理会抑制溶酶体功能,正如先前报道的那样。此外,JNK 激动剂 Anisomycin 联合自噬抑制剂巴弗洛霉素 A1 和氯喹,而非蛋白酶体抑制剂 MG132 和卡非佐米,恢复了 ZDHHC14 的水平,表明 JNK 介导的 ZDHHC14 降解主要受溶酶体降解途径调节(图 4f;补充图 S5e)。此外,Anisomycin 处理以剂量依赖性方式显著上调 ASCT2 水平,同时降低 ZDHHC14 水平(图 4g;补充图 S5f),而在 ASCT2C39S/C48S 突变体表达细胞中,ASCT2 的水平未受影响,表明 JNK 通路对 ZDHHC14 介导的 ASCT2 稳定性的影响确实依赖于棕榈酰化驱动的机制(图 4h;补充图 S5g)。与免疫印迹分析的观察结果一致,炔基-叠氮基点击化学实验结果表明,Anisomycin 显著抑制了 ASCT2 的棕榈酰化(图 4i, j),而 JNK 抑制剂在谷氨酰胺剥夺条件下则显著增强了 ASCT2 的棕榈酰化(补充图 S5h)。随后,研究者进行了Cycloheximide追踪实验,验证了 JNK 抑制剂在表达 ZDHHC14 的细胞中延长了 ZDHHC14 的半衰期并以时间依赖性方式促进了 ASCT2 的降解,而在 ZDHHC14 敲除细胞中,JNK 抑制剂仅导致轻微变化(图 4k, l)。因此,这些数据强调 JNK 通路是谷氨酰胺剥夺条件下调节 ZDHHC14 介导的 ASCT2 棕榈酰化的主要信号通路。
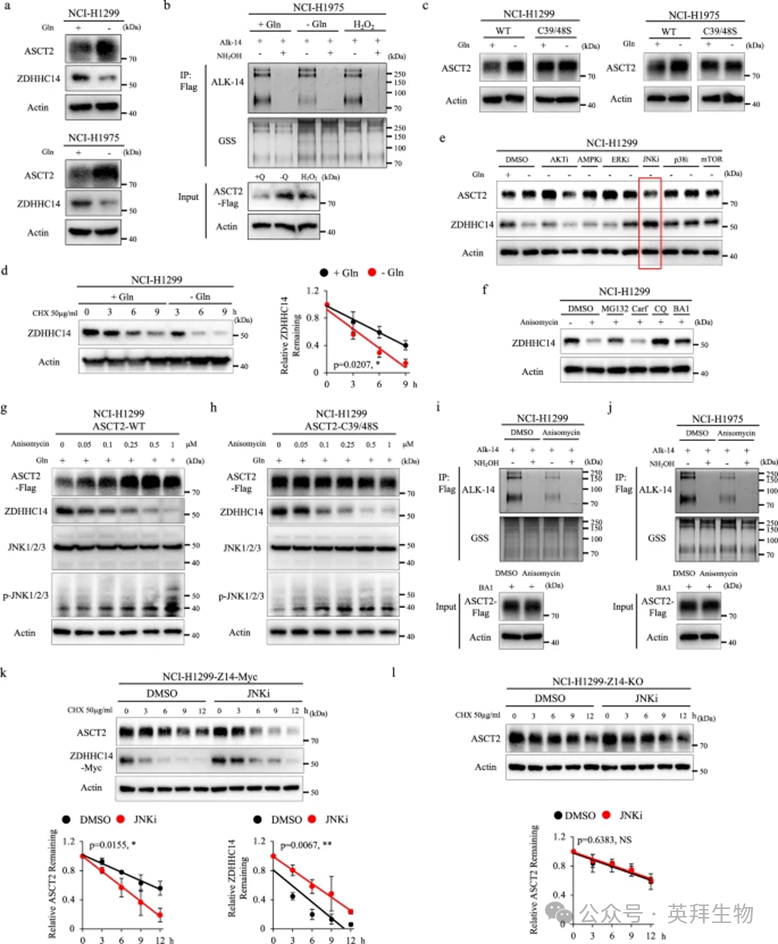
图4:谷氨酰胺剥夺通过JNK途径抑制ASCT2棕榈酰化
5 ZDHHC14响应谷氨酰胺剥夺刺激被JNK1在Thr440位点磷酸化
研究者接下来试图确定 JNK 调节 ZDHHC14 介导的 ASCT2 棕榈酰化的详细机制。JNK 通路包含三种异构体(JNK1、JNK2 和 JNK3)及其剪接变体,在细胞对各种应激信号的响应中起着关键作用。研究者通过免疫共沉淀分析确定了 ZDHHC14 与 JNK 异构体之间的相互作用,结果显示只有 JNK1 与 ZDHHC14 形成复合物(图 5a)。蛋白质-蛋白质对接分析也揭示了 ZDHHC14 和 JNK1 之间的蛋白质相互作用(补充图 S6a)。然后,研究者在 ZDHHC14 敲除细胞中过表达 ZDHHC14 以模拟野生型细胞系进行免疫共沉淀分析,结果显示 ZDHHC14 与 ASCT2 和 JNK1 都有强相互作用(补充图 S6b)。此外,JNK1 可以直接结合 GST-ZDHHC14 而非单独的 GST,这表明 JNK1 和 ZDHHC14 之间存在直接相互作用(图 5b)。为进一步明确 JNK1 对 ZDHHC14 的潜在影响,研究者将敲低 JNK1 表达的 NCI-H1299 和 NCI-H1975 细胞在含有或不含谷氨酰胺的培养基中培养,结果显示,在谷氨酰胺剥夺条件下,敲低 JNK1 显著提高了 ZDHHC14 的表达并抑制了 ASCT2 的表达(图 5c)。一致地,JNK-IN-8 和 CC-90001 处理(两者均抑制 JNK1 磷酸化和活化)在谷氨酰胺剥夺条件下导致 ZDHHC14 积累并降低 ASCT2 水平(图 5d)。
为了鉴定响应谷氨酰胺剥夺刺激而被磷酸化的 ZDHHC14 氨基酸残基,研究者建立了体外激酶实验并结合液相色谱-串联质谱分析(补充图 S6c)。有趣的是,在 ATP 刺激下,鉴定出 ZDHHC14 的三个磷酸化位点:Thr124、Thr440 和 Ser455(图 5e, f;补充图 S6c, d)。为了评估影响 ZDHHC14 蛋白稳定性的磷酸化位点,研究者在表达野生型 ZDHHC14 或非磷酸化突变体的细胞中检测了谷氨酰胺剥夺条件对 ZDHHC14 蛋白水平的影响。有趣的是,与野生型 ZDHHC14 相比,只有 ZDHHC14T440A 突变体在谷氨酰胺剥夺条件下显著恢复了 ZDHHC14 蛋白水平的降低,表明 Thr440 位点对于 ZDHHC14 蛋白稳定性至关重要(补充图 S6e)。此外,用Cycloheximide处理的表达野生型 ZDHHC14 或非磷酸化模拟 ZDHHC14 突变体的细胞显示,在谷氨酰胺剥夺条件下,ZDHHC14T440A 以时间依赖性方式延长了 ZDHHC14 蛋白的半衰期(图 5g;补充图 S6f)。一致地,野生型 ZDHHC14 而非 ZDHHC14T440A 突变体显著调节了响应谷氨酰胺剥夺的 ZDHHC14 和 ASCT2 的稳定性以及 ZDHHC14 的磷酸化,而这种调节效应被 JNK 抑制剂强烈抑制(图 5h;补充图 S7a, b)。从图 5i 可以看出,ZDHHC14T440A 也以剂量依赖性方式抑制了 Anisomycin 处理引起的 ZDHHC14 降解。最后,研究者进行了炔基-叠氮基点击化学实验,结果显示,在谷氨酰胺剥夺或 Anisomycin 处理后,ZDHHC14T440A 突变体表达显著恢复了 ASCT2 的棕榈酰化水平(图 5j;补充图 S7c, d)。总之,这些数据证明,响应谷氨酰胺剥夺,JNK1 被激活,进而磷酸化 ZDHHC14 的 Thr440 残基,引发 ZDHHC14 降解,从而抑制 ASCT2 的棕榈酰化修饰,并随后导致 ASCT2 积累增加。
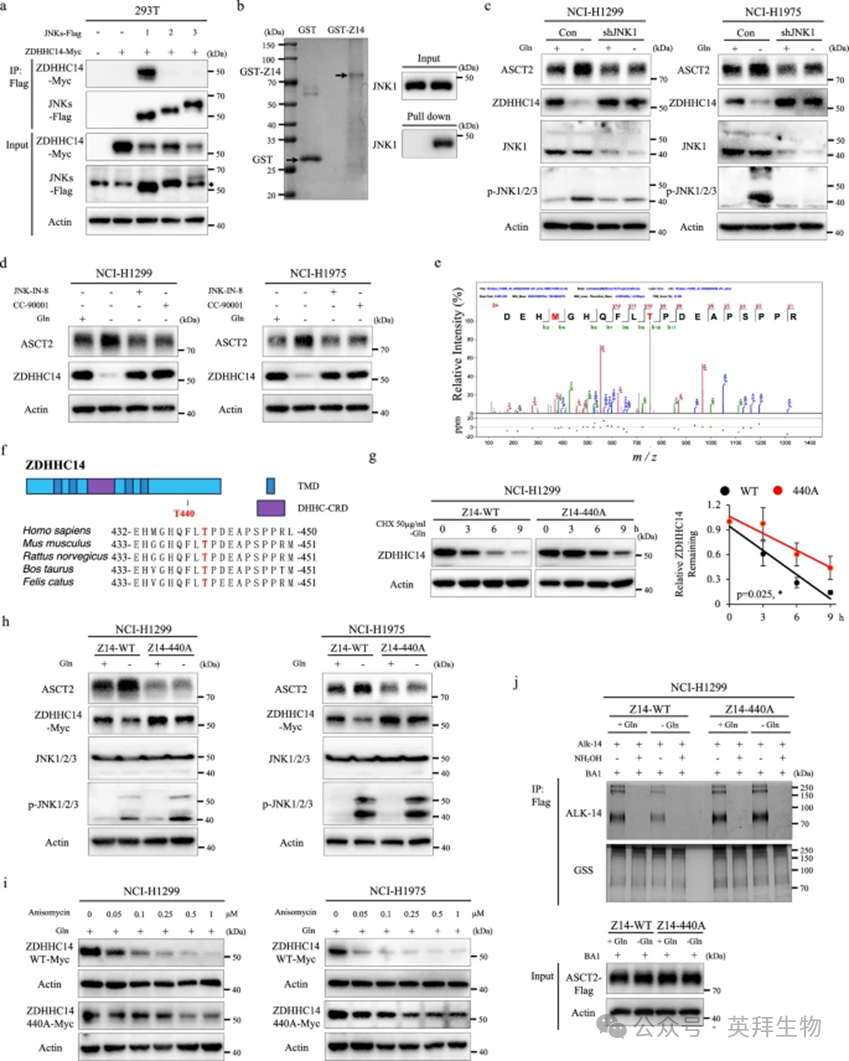
图5:JNK1介导的磷酸化通过触发ZDHHC14降解稳定ASCT2
6 ZDHHC14介导的ASCT2棕榈酰化调节谷氨酰胺代谢和肿瘤发生
ASCT2 作为一种谷氨酰胺转运蛋白,控制细胞对谷氨酰胺的摄取,并参与肿瘤和代谢疾病的进展。因此,研究者进行了谷氨酰胺摄取实验和谷氨酸生成实验,以进一步确定 ASCT2 棕榈酰化的代谢变化。正如预期,ZDHHC14 显著抑制了谷氨酰胺摄取和谷氨酸生成,而催化失活的 ZDHHC14 突变体(ZDHHC14C195S)在 NCI-H1299 和 NCI-H1975 细胞中仅引起微小变化(图 6a)。这些结果表明,ZDHHC14 作为 ASCT2 棕榈酰化的棕榈酰基转移酶,是谷氨酰胺代谢的重要因素。
在确定了 ZDHHC14 与非小细胞肺癌代谢改变的关键关联后,研究者试图评估 ZDHHC14 在体外和体内的功能作用。研究者首先通过生长和集落形成分析检测了体外增殖情况,结果显示,野生型 ZDHHC14 显著抑制了 NCI-H1299 和 NCI-H1975 细胞的细胞生长和集落形成,而催化失活的 ZDHHC14 突变体(ZDHHC14C195S)仅引起微小影响(图 6b-d)。通过肿瘤异种移植实验在体内也获得了类似结果,实验中将稳定表达空载体、野生型 ZDHHC14 或催化失活 ZDHHC14 突变体的 NCI-H1299 细胞注射到雄性 BALB/c 裸鼠体内。如图 6e-g 所示,注射稳定表达野生型 ZDHHC14(而非催化失活 ZDHHC14 突变体)的 NCI-H1299 细胞,在体内显著抑制了肿瘤生长。为进一步评估 ZDHHC14 介导的 ASCT2 Cys39 和 Cys48 棕榈酰化对肿瘤发生的影响,将共表达 ZDHHC14 和野生型 ASCT2 或 ASCT2C39S/C48S 突变体的 NCI-H1299 细胞皮下注射到裸鼠体内。正如预期,与表达野生型 ASCT2 的细胞相比,表达 ASCT2C39S/C48S 突变体的细胞表现出显著增强的肿瘤生长。值得注意的是,ZDHHC14 表达仅抑制了野生型 ASCT2 表达组的肿瘤生长,而对 ASCT2C39S/C48S 突变体表达组没有影响(图 6h-j)。
鉴于 ZDHHC14 介导的棕榈酰化在非小细胞肺癌中的关键功能,研究者假设 ZDHHC14 和 ASCT2 的表达与临床数据集中患者的生存率相关。支持这一假设的是,临床数据分析显示,与正常组织相比,非小细胞肺癌组织中 ZDHHC14 的 mRNA 水平显著降低(图 6k;补充图 S8a)。此外,Kaplan-Meier 图数据库显示,ZDHHC14 表达水平较低的非小细胞肺癌患者生存期较短(补充图 S8b)。相反,ASCT2 的高表达与非小细胞肺癌密切相关,并与较差的总生存期相关(图 6k;补充图 S8a-c)。与生物信息学数据分析一致,免疫组织化学染色分析进一步证实 ASCT2 和 ZDHHC14 之间存在负相关性(图 6l;补充图 S8d)。总之,这些分析揭示,高水平的 ASCT2 表达与 ZDHHC14 表达负相关,并且对非小细胞肺癌的临床进展至关重要。
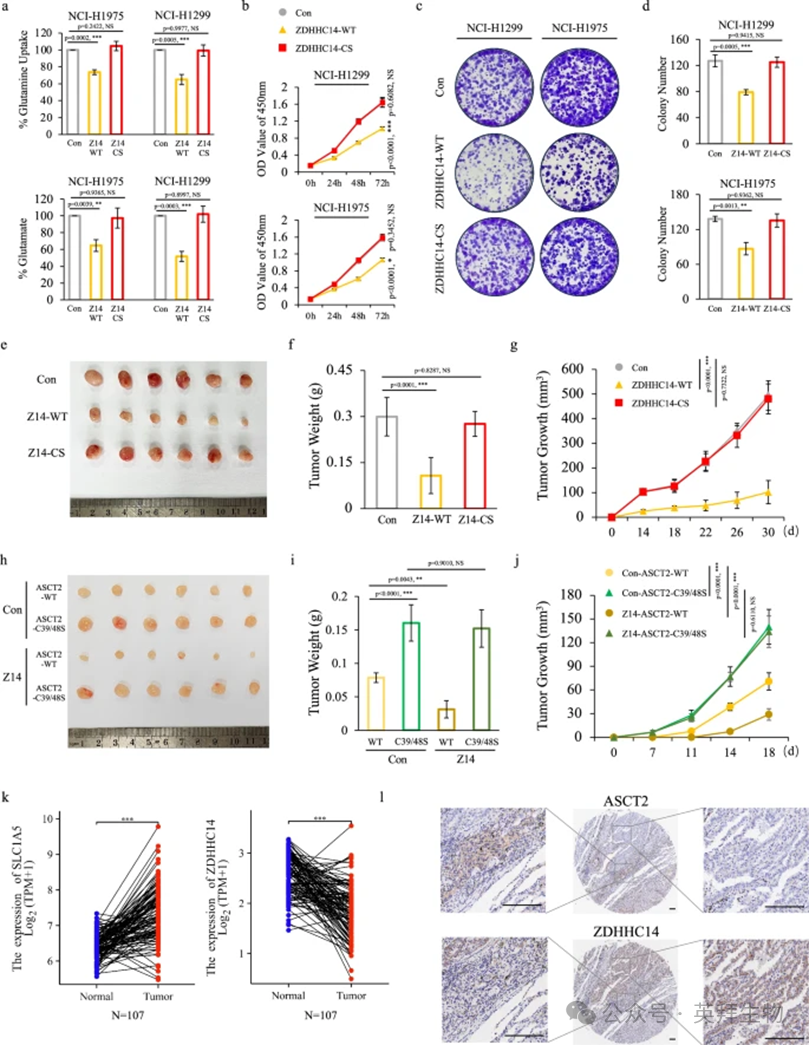
图6:ZDHHC14-ASCT2轴调控非小细胞肺癌中的谷氨酰胺代谢和肿瘤发生
7 靶向JNK通路增强ASCT2抑制剂V9302的抗癌效果
最近的研究表明,用 V9302 药理学阻断 ASCT2 可减弱癌细胞的生长和增殖。鉴于研究者观察到 JNK1 磷酸化 ZDHHC14 以促进其降解,从而增强棕榈酰化介导的 ASCT2 稳定性,研究者假设靶向 JNK 通路可能与抗 ASCT2 疗法产生协同作用,从而增强治疗效果。研究者首先确定 JNK 激活是否导致谷氨酰胺代谢发生变化。正如预期,JNK 激动剂 Anisomycin 在内源性表达野生型 ASCT2 的细胞中显著促进了谷氨酰胺代谢,但在表达 ASCT2C39S/C48S 突变体的细胞中则无此作用(图 7a, b),进一步支持 JNK1 通过介导 ASCT2 棕榈酰化来调节谷氨酰胺代谢。同时,基于 ABHD17B 与 ASCT2 棕榈酰化之间的调节关系,研究者还用 ABHD17 抑制剂 ABD957 处理细胞,并进行谷氨酰胺摄取和谷氨酸生成实验,结果显示 ABD957 显著抑制了 NCI-H1299 和 NCI-H1975 细胞中的谷氨酰胺代谢(补充图 S9a, b)。随后,JNK 抑制剂与 ASCT2 抑制剂的联合使用协同抑制了谷氨酰胺摄取和谷氨酸生成(补充图 S9c, d)。
为了进一步确定 JNK 介导的 ASCT2 棕榈酰化的功能后果,研究者测量了 JNK 抑制剂 JNK-IN-8 在 NCI-H1299 和 NCI-H1975 细胞中的 IC50 值,并使用低毒的 IC10 值来确定对 V9302 的增敏作用(补充图 S9e, f)。如图 7c, d 所示,JNK-IN-8 在所测试的浓度下增加了 NCI-H1299 和 NCI-H1975 细胞对 V9302 的敏感性。此外,集落形成分析显示,JNK 抑制剂 JNK-IN-8 和 ASCT2 抑制剂 V9302 的联合使用比单独使用任一化合物对集落形成产生了更强的抑制作用(图 7e, f)。
为了评估这些抑制剂在体内的抗癌效果,研究者将 NCI-H1299 细胞注射到 Balb/c 裸鼠体内,随后评估肿瘤对单独 JNK-IN-8 治疗、单独 V9302 治疗或两种药物联合治疗的反应。NCI-H1299 细胞接种两周后,当肿瘤体积达到约 100 mm³时,将小鼠随机分配接受以下治疗:(1)溶剂对照,(2)JNK-IN-8(20 mg/kg/天;腹腔注射),(3)V9302(20 mg/kg/天;腹腔注射),(4)JNK-IN-8(20 mg/kg/天;腹腔注射)和 V9302(20 mg/kg/天;腹腔注射)联合治疗,持续 16 天(图 7g)。与研究者之前在集落形成分析中的结果一致,JNK 和 ASCT2 抑制剂联合治疗组的抗肿瘤效果优于单一疗法(图 7h-j;补充图 S9g)。免疫组织化学染色分析进一步证实了 ASCT2、磷酸化 JNK 和 ZDHHC14 之间的强关联性(图 7k;补充图 S8d)。总之,这些分析揭示,靶向 JNK 和 ASCT2 的双重药物联合治疗在体外和体内对抑制致瘤性具有协同作用,为非小细胞肺癌的临床治疗确定了一种合理的药物组合。
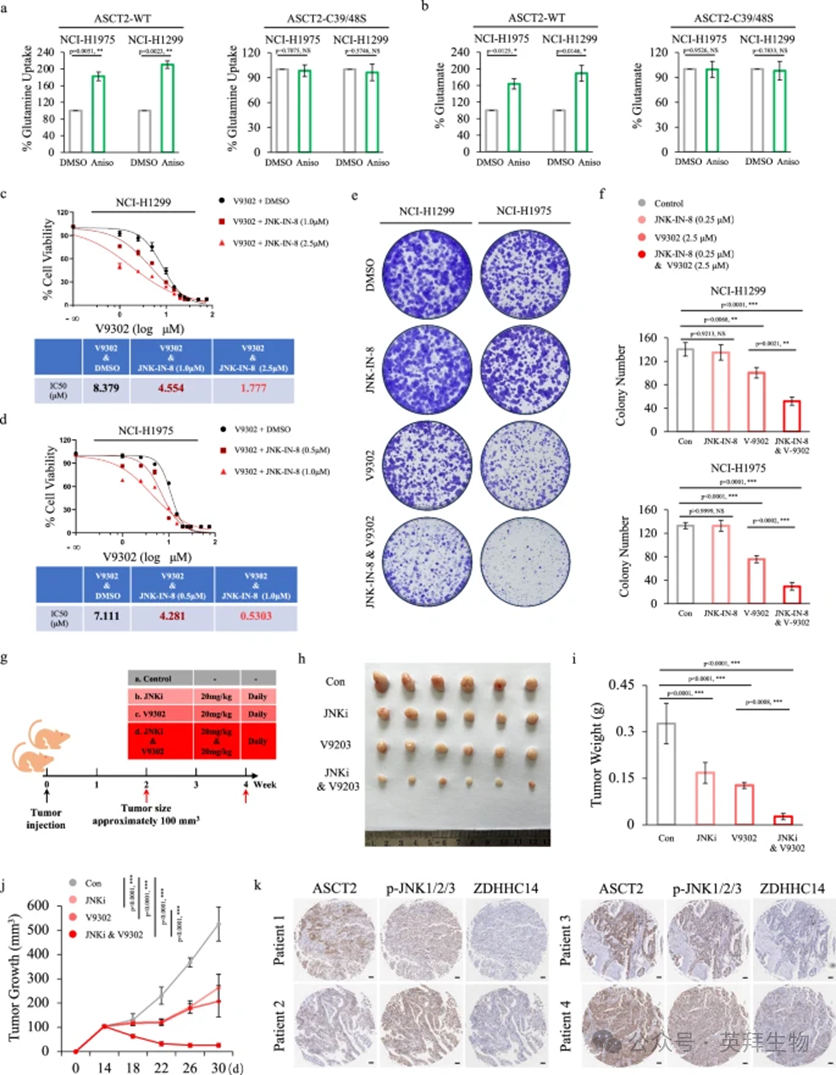
图7:V9302与JNKi联合组合在NSCLC中的协同抗癌活性
结论:
总之,研究者的研究揭示了一种连接非小细胞肺癌中营养感知与谷氨酰胺转运的新型磷酸化-棕榈酰化级联反应。研究者展示了 JNK1 介导的 ZDHHC14 磷酸化如何控制 ASCT2 的棕榈酰化和稳定性,揭示了一种代谢适应机制。这些发现确立了棕榈酰化作为癌症代谢中的一个关键调控节点,并强调了 JNK1-ZDHHC14-ASCT2 轴作为一个潜在的治疗靶点。
参考文献:
Chen X, Ke Z, Wei S, Chen J, Zhu K, Xu J, Zhao Y, Cen M, Jin Y, Pan Z, Xiong J, Chen Y, Dong C, Cao Q, Cao C. ASCT2 palmitoylation regulated by JNK1-ZDHHC14 axis orchestrates glutamine metabolism and NSCLC progression. Cell Discov. 2026 Feb 24;12(1):13. doi: 10.1038/s41421-026-00870-z. PMID: 41730846; PMCID: PMC12929575.